Drugs and their Receptors
General and local anesthetics
Pharmacology looks at the effect of drugs on changes in the physiology of the organism. These effects can be described without any knowledge of a receptor structure or function. More importantly, pharmacology includes all aspect of drug absorption, metabolism, and clearance, the potency, side effects, and compatibility with other drugs administered at the same time. It is not uncommon to find patients in hospitals that are treated with as many a eight substances. The combination of drugs can change the way the affect the physiology and this aspect is also important for anaesthesioloists.
General anaesthesia induces reversible depression of consciousness. Substances that induce general anaesthesia are applied in high concentrations and delivered to all parts of the body. They act on neuromuscular junction and heart muscle at the peripheral nervous system and in the central nervous system (CNS). An example of general anaesthetica are the opiates. Their effectiveness as a drug, of course, has been established long before any mechanism had been established. Today we know structure and function of opiate receptors, initially identified as naloxone binding sites.
General anaesthetics include a very broad spectrum of molecules and no particular chemical structure is associated with their effectiveness, except for the general hydrophobicity. It has been found that lipid soluble drugs tend to be more potent enzyme inhibitors than water soluble ones. This is true for general anaesthetics, which show a correlation between lipid solubility and effect. The higher their hydrophobic characteristic, the more potent as anaesthetica.
Fig. Correlation of drug potency and lipid solubility
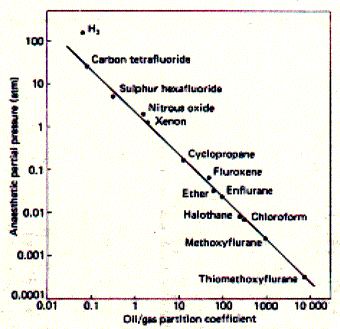
Potency is measured a partial pressure which is different
for different anaesthetica because of their different volatility. Weak
anaesthetica need to be applied at high partial pressure in blood to be
effective.
General anaesthetics range from simple gases like Xenon to complex structures of steroids. A widely used general anaesthetic is halothane which shows a similar potency as chloroform. Both substances are highly halogenated carbon units. The structure of halothane is shown below:
Fig. Halothane
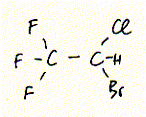
Although the mechanism for some general anaesthetics is known, this is not true for most of them. They share, however, some non-specific aspects in their effectiveness. General anaesthetics bind to a non specific locus which is identified as the cell membranes of excitable cells, i.e., cells of the nervous system and muscle cells. These membranes contain ion channels and receptors which are the primary target of many drugs including general and local anaesthetics. This non specificity for membrane binding explains in part while there is no known antagonist to general anaesthesia. The membrane hypothesis is supported by some models which explain the mechanism in some or other ways as membrane expansion and disordering mechanism. In other words, general anaesthetics may work through their affect on the fluidity of the phospholipid bilayer which in turn affects the activity of the membrane proteins responsible for action potentials.
In contrast, local anaesthetics, which are used to only locally suppress neuronal activity without the loss of overall consciousness, have been shown to inhibit excitation-conduction processes of peripheral nerve fibers and endings. The cocaine and lidocaine family of local anaesthetics are well described channel blockers, which in contrast to lipid soluble general anaesthetics, do not partition into the membrane to interfere with protein activity, but directly diffuse into the open conformations of ion channels thereby blocking ion flux and thus excitability.
Fig. Mechanism of binding to ion channels and receptors of local
and general anaesthetics
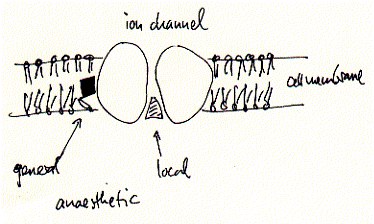
The effectiveness of local anaesthetics is concentration dependent. For
the lidocaine and cocaine family of drugs the following minimum blocking
concentrations Cm have been established:
| local anaesthetic | Cm (mM) |
| procaine | 8.0 |
| lidocaine | 2.5 |
| cocaine | 1.5 |
| tetracaine | 0.6 |
| dibucaine | 0.2 |
Electrophysiology as tool to study
drug-receptor interaction
The mechanisms of drug-receptor interaction has been elucidated by combining structural studies with electrophysiological experiments. The latter implores the 'enzyme' kinetics of ion channels and with the development of patch clamp technique provides an extremely sensitive monitoring assay down to the level of a single active receptor unit.
Electrophysiology assess the transport rates of ions through ion channels. This transport is measured as current, or ionic current, because ions carry charges. Thus, the flux of ions across membranes can be compared to a simple electronic circuit composed of a ohmic resistor Rm -- the ion channel -- in parallel to a capacitor, Cm, -- the phospholipid bilayer, and 'battery', or transmembrane potential Vm, due to asymmetric distribution of ions, i.e., ion gradients.
Fig. Equivalent circuit of a membrane-ion channel system
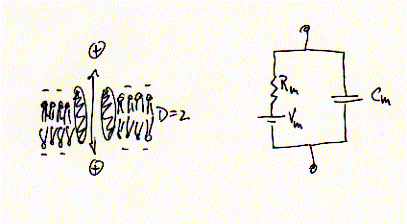
While ions (+) can pass freely across the ion channel, they are prohibited in doing so across the los dielectric medium (D=2) of the membrane. Ion currents can be measured in very different membrane systems. Two in vitro systems shall be described here in more detail; the Xenopus frog oocyte voltage clamp and the planar lipid bilayer.
Oocyte two electrode voltage clamp
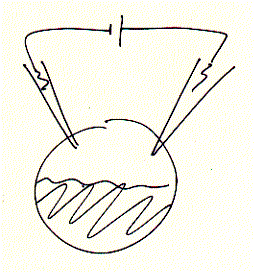
Many ion channels are difficult to isolate and purify because they are found in small copy numbers and in membrane areas mixed with many different types of channels and receptors. Molecular biology techniques allowed to clone and express a desired protein in heterologous expression systems, i.e., in cells where they are usually not expressed and under conditions where very large quantities of the protein can be produced (over expression). An ideal system for electrophysiology has been found in unfertilized frog oocytes. When injecting mRNA synthesized in vitro (or extracted from cells, as mixture though) the egg synthesizes protein from the injected mRNA template in copy numbers exceeding manifold the concentration of endogenous levels of the same or similar types of proteins of the oocyte itself. Thus, large signals can be generated and clearly distinguished as being the product of the injected mRNA. Two electrode voltage clamp assays are used to study macroscopic membrane currents. Here, two microelectrodes are inserted into a single frog egg, with one electrode controlling the membrane potential, while the second electrode measures the current flowing across the entire oocyte membrane.
Fig. Xenopus oocyte two electrode voltage clamp and elicited Na+
inward current of expressed serotonin receptor
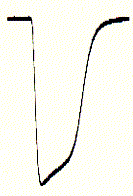
inward currents (right) are seen as downward signals (negative
current; for convention on voltage and current polarity see text), mRNA
coding for the ionotropic serotonin receptor (5HT-3A) subtype has been
injected one day prior to the voltage clamp experiments. The membrane
is held constant (clamped) at -70mV (inside negative) and 10mM
serotonin (agonist) added to the bath solution surrounding the oocyte.
An immediate downward current could be observed which shows a biphasic
decay (inactivation) shortly after;
Oocyte two electrode voltage clamp has been successfully used for many
different types of ion channels. The ease of RNA injection and the large
size of the cell makes it suitable for handling and generating a lot of
data in a very short time. 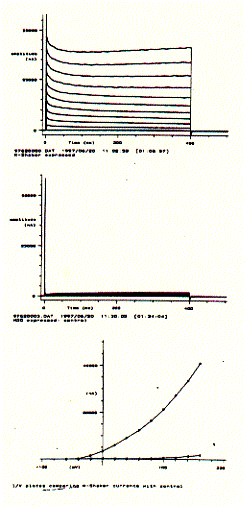 By
analyzing the current amplitude and time course of the macroscopic current
as shown above as a function of membrane potential (voltage), ion concentration,
or agonist concentration and type, a detailed structure function relationship
of the expressed membrane protein (complex) will be obtained.
By
analyzing the current amplitude and time course of the macroscopic current
as shown above as a function of membrane potential (voltage), ion concentration,
or agonist concentration and type, a detailed structure function relationship
of the expressed membrane protein (complex) will be obtained.
For voltage gated ion channels like neuronal potassium selective ion channels, current recordings can be generated in 'bundles', where each current trace corresponds to a specific voltage clamp period. By cycling through a resting period and incremental increases in membrane potentials, current/voltage curves can be generated demonstrating the voltage dependence of an ion channel protein complex.
The figure to the left shows a Drosophila Shaker K-channel experiment in a single Xenopus oocyte. The upper panel shows a bundle of macroscopic outward current traces showing the very fast rising phase (voltage activation) and slow and impartial decrease or inactivation. The middle panel shows the same voltage protocol for a non-injected oocyte which subsequently does not express any Shaker K-channel. It does, however, show a small endogenous current component for very large, positive membrane potentials. Plotting the end currents (bottom panel; where the voltage is switched back to its resting level of -80mV) against the applied voltages (going from -60mV to +160mV in 20mV increments) shows that this particular potassium channel is closed at negative membrane potentials and can be activated (channel opens) at membrane potential more positive than -40mV. The current/voltage diagram also shows that while the oocyte with injected 5HT-3 mRNA shows large currents at positive membrane potentials, the non-injected cell shows a small endogenous current at voltages above +100mV. This endogenous current, of course, contributes to the total membrane current of injected oocytes and can be subtracted electronically, if necessary, to extract the pure K-channel current activity.
Having established the intrinsic activity of an ion channel, agonists, antagonists, and channel blockers can be tested for their effectiveness on modulating channel function. Two K-channel blockers, tetraethylammonium (TEA) and charybtotoxin (CTX) can be used to further characterize the currents measured of 5HT-3 mRNA injected oocytes. This pharmacological profiles unequivocally demonstrate the presence of a particular subtype of an ion channel or receptor.
Fig. Shaker K-channel inhibition by TEA and CTX
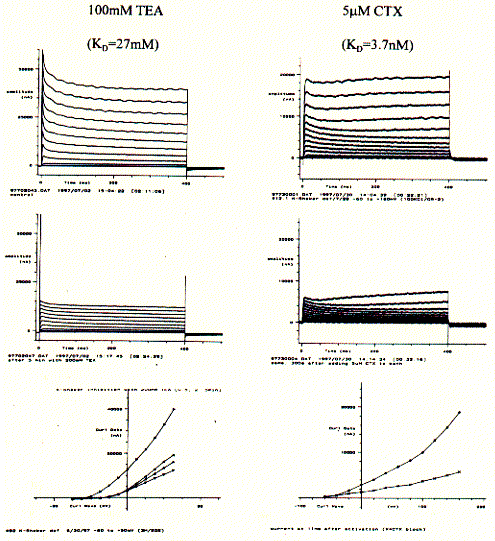
Similar to the current recordings and current/voltage curves shown for
above, the addition of K-channel specific blockers show a decrease in
membrane current. The KD numbers indicated the affinity with
a lower number representing a higher affinity of better binding of the
blocker with the channel protein. The snake venom CTX is a highly potent
toxin and demonstrably interferes with neuronal activity by selectively
blocking K-channel activity. The less potent tetraethyl ammonium salt
TEA also is specific for K-channels, but needs be applied in much higher
concentration to block fifty percent of the channel population in an experimental
system (this is due to the proper definition of the dissociation constant
KD). While CTX binds to the extracellular surface of the channel,
TEA has to cross the cell membrane and is effective only if it can bind
to the cytoplasmic entrance of the pore.
Single channel recordings in planar lipid bilayers
When channels or receptors can be isolated and purified, it is often possible to reconstitute them into an synthetic membrane system which is composend of phospholipids and only one type of protein. Such a system, similar to patch clamp experiments on cell membranes (a mujch more sensitive current detection method than the two electrod voltage clamp technique), allows to measure currents through single ion channel units. The recordings obtained from are called single channel recordings.
Fig. Bilayer formation on glass micropipette tip and single channel
recording
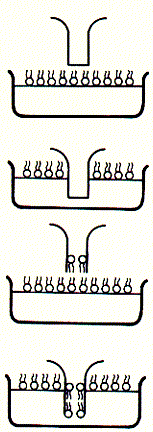
Note: pipette tip is inserted twice in to the bath solution
througha lipid monolayer; during the two step procedure, a single bilayer
forms across the buffer filled tip of the glass opening (bottom); after
inserting an ion channel into the bilayer covering the pipette tip, current
recordings like that to the right can be observed; here, the current traces
are continous and show two levels; an upper level ( in top trace) corresponds
to the open state of the channel, while the lower level represents the
closed channel; note that channel open and closed times can vary substantially;
channel open to closed transition can occur in rapid succession;
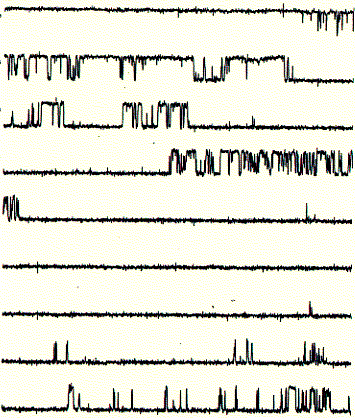
These single channel recordings (one closed and one open level at any given time during the recording) are due to the presence of only one active channel unit. In the recording shown above, the open probability (time spent in the open state divided by the total time of recording) is above 40%. If the membrane would contain more than one active channel unit, it would be very likely to observe occasional opening of a second level on top of the first open level. Thus, the interpretation here is that the current shows the activity of a single channel unit. From the recording it cannot be concluded how many inactive or closed channel units are present in the same membrane.
Fig. Single channel analysis of current amplitude (left) and open
dwell time distribution (bottom)
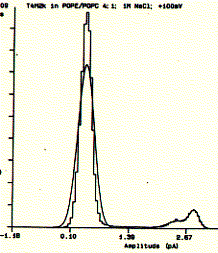
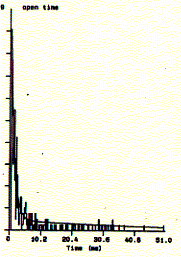
Note: left panel: large peak corresponds to closed level
at 0.3pA and small peak to open state at 2.6pA; right panel: open dwell
time distribution in milliseconds;
Local anaesthetics in reconsituted membrane systems
The planar bilayer system is particulary suitable to study structure function relationship of local anaestethic interaction with ion channels. The latter can be purified to homogeneity of replaced by synthetic peptide systems mimicking the structure of the authentic ion channel structure. The latter approach shall be described here.
Using the tip-dip technique, proteins or peptide can be inserted into preformed membranes made of synthetic phospholipids. Thes assures highest purity of the system and allows interpretation of experimental results for intrinsic channel properties. These are poperties which appear to be determined only by the protein (peptide) structure itself, immersed in a phospholipid bilayer.

Here, protein detergent extracts or lipid vesicle containing protein can be added to the subphase of the bilayer (bathsolution outside the glass pipette tip). Both methods yield spontaneous incorporation of protein into the preformed lipid membrane or fusion of the added vesicle membranes with the planar membrane on the pipette tip.
A synthetic peptide channel approach makes use of alpha helical bundles which are tethered by a short turn forming peptide.
Fig. Stereo view of a 4 alpha helical bundel peptide (design by
M.Mutter)
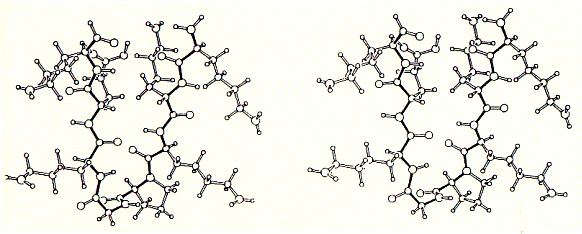
The final structure is represented as a cartoon. The horseshooe like
nona-peptide contains four lysine residues.  A
single alpha helix is synthesized to the side chain NH2 group forming
four parallel arranged peptides with equal sequence. The amino acid sequences
can be choosen at will and are representing the channel ligning transmembrane
sequences of the nicotinic acetylcholine receptor (M2) or Ca selective,
voltage gated ion channel subunit alpha.
A
single alpha helix is synthesized to the side chain NH2 group forming
four parallel arranged peptides with equal sequence. The amino acid sequences
can be choosen at will and are representing the channel ligning transmembrane
sequences of the nicotinic acetylcholine receptor (M2) or Ca selective,
voltage gated ion channel subunit alpha.
Initial experiments of synthetic peptides which were not covalently liked clearly showed ion channel activity in planar membranes, although the current amplitutes proved to be heterogenous reflecting the intrinsic variality of such a self-assembly channel system. In the covalently linked 4-helical bundle arrangement, a single unitary conductance could be ovbserved representing the ion channel formed by the four alpha helical transmembrane peptides.
Comparing channel forming properties from peptides with many different sequences, the amphipathic characteristic of the alpha helix proved to be important, because entirely hydrophobic peptides did not induce any channel like activity, also the membrane stability was compromized indicating peptide membrane interaction. Naturally occuring peptides with many positively charged amino acid residues (magaining peptides from frog skin) are strong antibiotic agents and show membrane disruptive activity rather than controlled stable ion channel activity.
Fig. Frog skin antibiotic magaining 2 shows typical
membrane disruptive activity
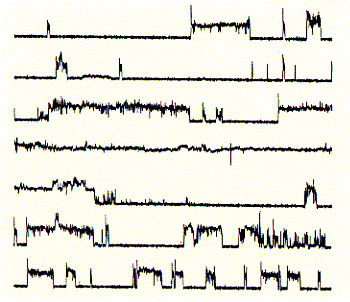
Note: continious recording in planar membrane at +100mV;
note
the less stable open current levels as compared to regular
ion
channel activities;
Having established an experimental ion channel assay systems allows the
measurement of local anaesthetic activity. 4-helical bundle peptides with
the sequence from the delta subunit of the muscular nicotinc acetylcholine
receptor protein (M2d) can be blocked by the
lidocaine derivative QX-222 in the same way as the native receptor channel.
Fig. QX-222 block of synthetic peptide channel
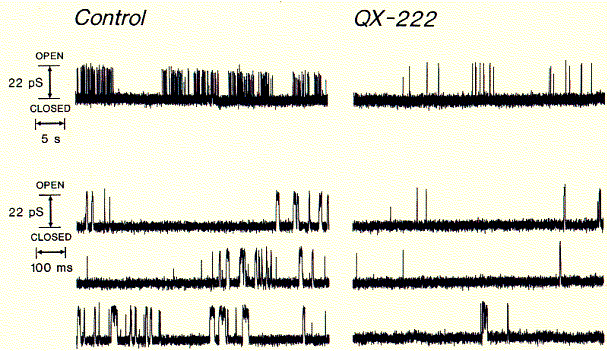
Comparing the left (control) current recording with one in the presence of channel blocker clearly shows the decrease in opening events (upward deflections). This is due to the inhibitor binding inside the open channel conformation blocking ion flux across the membrane. The binding of lidocaine derivatives inside the pores of cation selective ion channels has been demonstrated by measuring an increased blocking effect when the channels are in an activated mode, wherease the blocker has no effect, when applied to the closed channels and washed off before the pores are opened again. Also biochemical cross linking studies such as those with the general channel inhibitor chlorpromazine (CPZ) demonstated that the molecule interacts with amino acid residues pointing toware the center of the conducting pore structure.
Channel inhibitors used to treat hear beat irregularities or anxiety attacks like the diazepam family of drugs, including the derivative BayK-8644, act through binding sites on the protein surface exposed to the lipid phase of the membrane. Comparing the chemical structures of QX222 and BayK-8644 shows that while the former is amphipathic and sits inside the hydrophilic pore, attracted by its the positive charge to the negative residues of the pore lining structure, the hydrophobic diazepam incorporated into the membrane and diffuses towards channel surface binding sites, thereby allosterically and closing the channel gate.
Fig. Calcium channel blocker (+) BayK8644 and local anaesthetic
QX-222, a lidocaine derivative
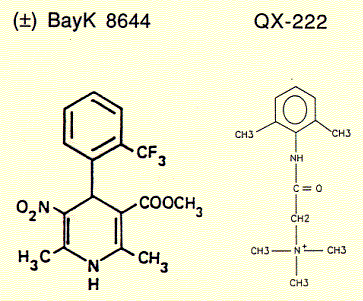
BayK 8644 is a enantiomeric structure, meaning it exists in two chemically equivalent stereoisomers, + and -. It can be shown that only the (+) enantiomer is a potent blocker of calcium channels while the (-) isoform is a well known agonist. Surprisingly, this stereospecific behavior is retained for the synthetic channel mimicking peptide bundle, although the four helices in this synthetic system are not supposed to be exposed to the active substance. There is, however, a possibility that the active component is able to diffuse into the alpha subunit of those calcium channels directly interacting with the channel lining transmembrane segment, the sequence of which has been used to study the innermost channel module structure.
Fig. Stereospecific inhibition/activation of calcium channel mimicking
synthetic peptides
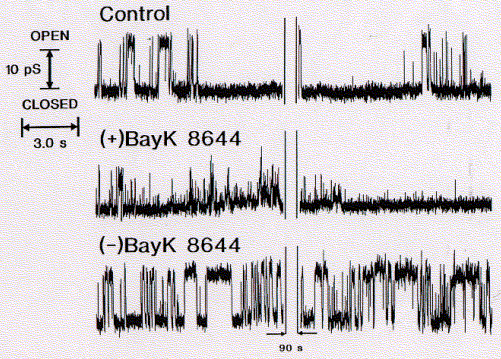
(data from A.Grove, UCSD)
In search of new drugs - the ionotropic
receptor model
An Introduction to the biology of the serotonin receptor subtype 5-HT3
Most targets of pharmacologically active drugs are ion channels and receptors. The development of pharmacaphore concepts has been of tremendous value for the identification and design of new agonists and antagonists for these receptors. These receptors come in two classes -- ionotropic and metabotropic. They are either ligand gated ion channels or G-protein coupled receptors that bind ligands, but transmitt a conformational signal across the cell membrane activating a cytoplasmic process (metabolic control) or a secondary ion channel.
Here the ionotropic subtype of serotonin gated receptors shall be discussed. Serotonin receptors are also found to be either GPCRs or ligand gated ion channels. While seven GPCR types of serotoning receptors are known, only two subtypes of the ionotropic receptor have been described. Serotonin receptor 3A, or 5HT-3A, has been cloned and sequenced in the 1980s and only in early 1999, has a second subtype been identified (5HT-3B). The latter is a regulatory subunit able to modulate the intrinsic channel activity of the 3A subtype. The abiltiy of 5HT-3A subunits to form homooligomeric channels in oocyte expression systems has greatly helped in understanding the structure function relationship of this subtype, including a well documented pharmacology.
Comparing the function of 5HT-3A in oocytes with that of endogenous 5HT-3
recordings clearly indicates that the differences in channel properties
must be due to the presence of other subunits, similar to the multi subunit
family of all other types of ligand gated receptors, namely the nicotinic
acetylcholine receptors with 9 subtypes, and the glutamate receptors wity
5 subtypes.
Structure and Molecular Function
The 5-HT3 (serotonin) receptor subtype is unique among known monoamine receptors in that, rather than being a G-protein-coupled receptor, it forms a ligand-gated ion channel. The subunit has structural and functional similarities with nicotinic AChR, GABAergic and, other ligand gated ion channels (1) and the receptor has therefore been modeled as homopentameric protein complex with allosteric ligand binding sites. Heterologous expression in Xenopus oocytes showed a fast 5-HT induced activation (<2ms) and desensitization.
Fig. Activation of 5HT-3A receptor by serotonin
(5HT; 5-hydroxytryptamine) and agonist chlorophenylbiguanide

Note: downward currents reflect Na+ inward currents; the
activity profile of the current is specific for the two different agonists
indicating different activation/inactivation kinetics;
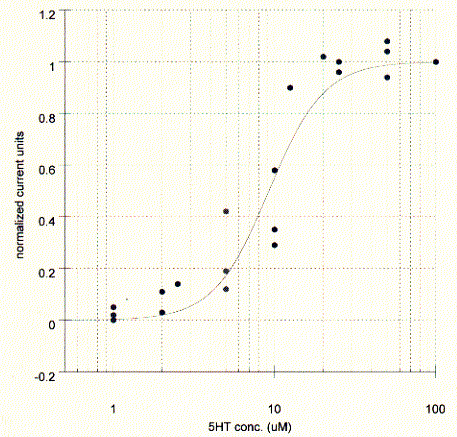
The elicited current is proportional to the agonist concentration and typical dose response curves can be measured by plotting the peak current against the agonist concentration. The affinity of the agonist for the binding site can be established as the concentration where the peak current is half maximal. The maximum current corresponds to the saturation level of the membrane, i.e, all present channels are activated, while at the midpoint concentration only 50% of all receptors are activated. The KD in this experiments is about 10mM for serotonin.
Fig. Serotonin dose response curve for 5HT-3A receptor in Xenopus
oocyte
The channel exhibited cation selectivity and a unitary conductance of 10-15pS. Current-voltage relationships did not show rectification in one report (2), but strong inward rectification for potentials more negative than -50mV in another report (3). The latter study estimates the single channel conductance at 0.4pS (from noise analysis of macroscopic courrents) which is about 20 to 100 fold lower than measured for other ligand gated receptor channels.
Fig. Inward currents in oocytes for NaCl, GuanidiniumCl, and CsCl
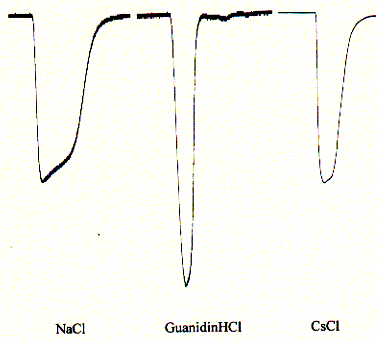
Note: the large peak for guanidinium chloride indicates
a preference for the quaternary ammonium ion guanidinium;
The 5HT-3A receptor is a cation selective channel (data not shown here) and is also weakly selective against divalent cations like calcium. While 2mM calcium reduces the current up to 50%, elevated (but physiologically unimportant) 10mM CaCl2 reduce the inward current by 90%. Since intracellular calcium concentrations are in the submicromolar range and extracellular concnetration abotu 1.5mM, calcium blockade is physiologically not relevant.
Fig. Calcium block of 5HT-3A receptor in Xenopus oocytes
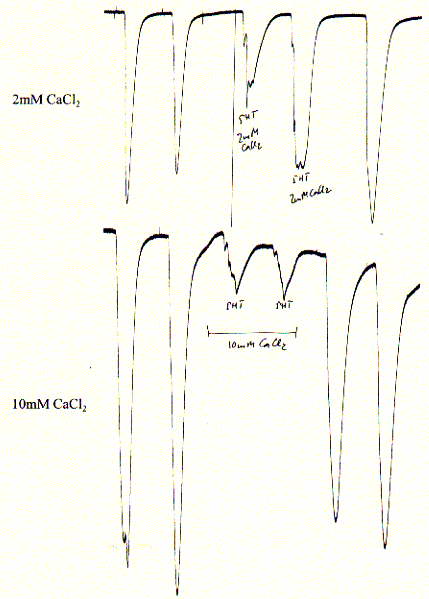
Note: experiments show two 5HT pulses before calcium addition
and two 5HT pulses after removal (wahsout) of calcium showing complete
restoration of initial activity and thus reversibility of blockade;
Two splice variants of the ligand-gated 5-HT3 receptor that differ
in a 6-amino-acid selection were cloned by polymerase chain reaction from
the hippocampus x neuroblastoma cell line HN9.10e. When expressed in Xenopus
oocytes, both variants individually formed 5-HT3 receptors showed idistinguishable
current responses to 5-HT and 1-phenylbiguanide activation and both variants
were equillay blocked by the specific antagonist LY 278584-maleate. For
both receptors, the monovalent cations Na+, K+,
Rb+ and Li+ showed the same relative permeability;
NH4+ showed a »
2.7 times higher permeability than Na+, whereas Tris+
was only poorly permeable. In contrast to other reports, the receptors
were completely and reversibly blocked by extracellular Cs+
in both oocytes and native HN9.10 cells. Moreover, Ca2+
exhibited a concentration-dependent decrease (0.9-18 mM) of 5-HT-induced
Na+ currents without affecting the inward rectification
of the current/voltage relation. The 2 receptors were reversibly inhibited
by nanomolar concnenctrations. of the specific inhibitors of protein kinase
C (PKC) bis-indolylmaleimide, but not by the equipotent and less specific
inhibitor staurosporine. A regulatory effect on both 5-HT3 receptor subunits
by PKC-mediated protein phosphorylation might be possible, however, a
functional role of the 2 splice variants present in 1 cell remains to
be determined (6).
Pharmacology
5-HT3 receptor antagonists have been shown to produce beneficial effects in animal models of cognitive and psychiatric disorders. Whether 5-HT3 receptor antagonists may have similar profound effects in the treatment of anxiety, depression or psychosis will be detd. by the outcome of ongoing clin. trials. However, it is in the treatment of cancer chemotherapy induced emesis that 5-HT3 receptor antagonists have had their greatest impact (1).
Serotonin (5-hydroxytryptamine or 5-HT) acts on 5-HT3 receptors in the central nervous system or on peripheral vagal afferent fibers to initiate vomit reflexes. 5-HT3 receptor antagonists block this action and thereby greatly reduce the no. of emetic episodes that occur during cancer chemotherapy. The marked clinincal efficacy of 5-HT3 receptor antagonists such as ondansetron, granisetron and tropisetron together with their lack of adverse side effects has greatly improved the treatment of cancer chemotherapy induced emesis (1). The cytotoxic agents used in cancer chemotherapy provoke the release of 5-HT from enterochromaffin cells in the gastrointestinal tract.
5-HT3 receptor-mediated ion currents evoked by the full agonists 5-hydroxytryptamine (5-HT), quaternary 5-HT (5-HTQ), meta-chlorophenylbiguanide (mCPBG) and the partial agonists dopamine and tryptamine have been investigated in whole-cell voltage clamp expts. on N1E-115 mouse neuroblastoma cells. All agonists desensitize the 5-HT3 receptor completely with a steep concentration. dependence and a potency order of: mCPBG > 5-HTQ » 5-HT .mchgt. tryptamine > dopamine. The time course of recovery from desensitization depends on the agonist used.
Fig. Dopamine as partial agonist of the 5HT-3A receptor

Note: dopamine induces small currents as compared to the
5HT-3 specific agonist chloro-phenylbiguanide; with dopamine concentrations
< 0.1mM no currents could be observed;
The affinities of drugs for both dopamine D2 and 5-hydroxytryptamine3 (5-HT3) receptor sites were determined in rat brain membranes. The 5 traditional antiemetics (chlorpromazine, prochlorperazine, droperidol, fluphenazine, and domperidone) displayed high affinity (<20 nM) for dopamine D2 receptors in corpus striatum but were inactive at 5-HT3 receptors in the cortex. In contrast, 5 recently developed 5-HT3 antagonists (BRL 43694, ICS 205-930, zacopride, Lilly 278584, and MDL 72222) displayed nanomolar affinity for the 5-HT3 site but were inactive (>10,000 nM) at the dopamine D2 receptor. Metoclopramide was unique among these agents in that it was similarly potent at dopamine D2 (240 nM) and 5-HT3 (120 nM) receptors (11).
Fig. Inactivation of 5HT-3A receptor in Xenopus oocytes by the
specific antagonist ICS 205-930
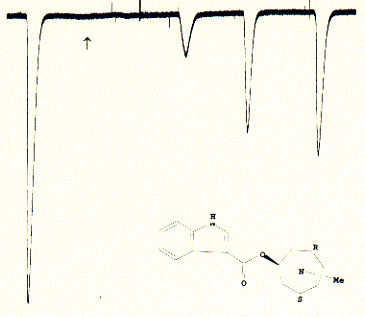
Note: this antagonist has a very high affinity for the 5HT3
receptor and does not wash out easily after administration; the slow and
incomplete increase of recovered currents is typical for these type of
strong binding;
A quantitative molecular model was derived to predict drug affinities for 5-hydroxytryptamine3 (5-HT3) receptors. The model was based on the molecular characteristics of a learning set of 40 pharmacological agents that had been analyzed previously in radioligand binding studies. Molecules were analyzed for various structural features, i.e., the presence of a benzenoid ring and nitrogen atom, substitutions on the benzenoid ring, the location of the substitutions on the nitrogen, and the molecular characteristics of the most direct pathway from the benzenoid ring to the nitrogen. Weighting factors, based on published 5-HT3 receptor affinity data, were then assigned to each of 10 molecular characteristics (17).
Fig. Pharmacophore structure identified by superimposing the six
core structures of high affinity compounds for the 5HT-3A receptor
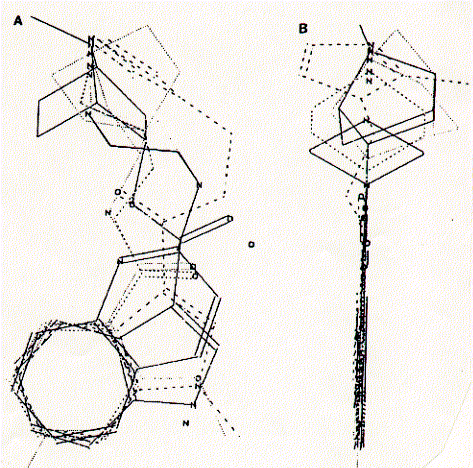
The composite structure is viewed (A) with the aromatic
ring in the plane of the figure and (B) with the aromatic ring placed
perpendicualr to the plane of the figure.
from Schmidt et al., 1989, Molecular Pharmacology 36:505-511.
The following nine rules have been established for the 5HT-3A receptor
pharmacophore:
1. contains aromatic ring structure (lower half of molecule; consistence with the hypothesis of Lloyd and Andrews which states that all central nervous system active drugs contain an aromatic ring; J.Med. Chem, 1986, 29:453-1093).2. a tropane ring embedded nitrogen is present (see upper half of molecule shown above) and located at a nearest distance from the aromatic ring is not more than seven atoms from the aromatic ring
3. when aligning the tropane ring nitrogen in the same plane as the aromatic ring (torsion angle flexibility) the distance between the nitrogen and the aromatic ring center is 6.0 to 7.8Å.
4. chemical substitutions of no more than 3 atoms are allowed at the nitrogen
5. the tropane ring structure itself does not tolerate substitutions larger than methyl groups (-CH3); larger groups significantly reduce affinity
6. the linker structure between aromatic ring and ring nitrogen contains steric similarities that reduces flexibility (carbonyls or C=O bonds)
7. the first and second atom from the aromatic ring in the linker is never a tetrahedral carbon (no torsion angle flexibility; see point 6)
8. the third atom from the aromatic ring may be a tetrahedral carbon
9. substitutions on the aromatic ring must be able to adopt a co-planar conformation
A naturally occuring drug, atropin, demonstrates how a 'small' but significant
violation of the 'rules' in its structure explains its very low affinity
for the serotonin receptor 3A subtype. The
first atom from the aromatic ring (lower left two bond structure) is a
tetragonal carbon and thus allows torsion anlge flexibility (see
rules 6 and 7) explaining that the structure can deviate from being co-planar
witht the aromatic ring. These two atoms are the main difference as compared
to the highly specific antagonist ICS 205-930. The molecular weight and
chemical formula are nearly identical although the affinity of atropin
for the 5HT-3A receptor is x1000 lower than that for ICS 205 930. Atropin
is a natural antagonist of cholinergic receptors (acetylcholine receptors).
The
first atom from the aromatic ring (lower left two bond structure) is a
tetragonal carbon and thus allows torsion anlge flexibility (see
rules 6 and 7) explaining that the structure can deviate from being co-planar
witht the aromatic ring. These two atoms are the main difference as compared
to the highly specific antagonist ICS 205-930. The molecular weight and
chemical formula are nearly identical although the affinity of atropin
for the 5HT-3A receptor is x1000 lower than that for ICS 205 930. Atropin
is a natural antagonist of cholinergic receptors (acetylcholine receptors).
The case of atropin and the 5HT-3A pharmacophore structure requirements
demonstrate the importance of molecula motion in structures. The
flexibility of the atropin molecule between aromatic and tropane ring
structures essentially reduces the chance in atropin of superimposing
the tropane ring and aromatic ring, a gross-structure reqirement for 5HT-3A
antagonist binding. Further structure-activity relationship studies show
that all known receptor antagonist show at least one degree of freedom.
This hinders potential screening of ligand-receptor topoplogy which is
best achieved with a rigid molecule as template for rational drug design.
This structural mobility is further demonstrated in thermodynamic analysis
of drug binding.
Agonist vs. Antagonist binding: understanding ligand binding mechanisms
Thermodynamic parameters DG°
(free energy), DH°
(enthlpy = heat) and DS°
(entropy = disorder) of the binding of agonists and antagonists to the
serotonin 5-HT3 receptor subtype have been determined by affinity measurements
carried out on rat cortex membranes at six different temps. (0, 10, 20,
25, 30, 35°C) and van't Hoff plots.
Affinity constants were obtained by displacement in inhibition assays
for the other compounds. Van't Hoff plots were essentially linear in the
temp. range investigated, showing that the heat capacity DCp°
of the binding equiliebrium is nearly zero indicating the absence of important
changes is intramolecular interactions or binding energies (potential
energy of non covalent bonds). Thermodynamic parameters are in the range
18 £DH°£
53 kJ mol-1 and 202 £DS°£
320 J K-1 mol-1 for agonists and -16 £DH°£
0 kJ mol-1 and 70 £DS°£
179 J K-1 mol-1 for antagonists indicating that agonistic binding is totally
entropy-driven while antagonistic binding is relatively less entropy-
and more enthalpy-driven. In the -TDS°
vs. DH°
plot the thermodynamic values are clearly arranged in distictly different
groups for agonists and antagonists, which, therefore, turn out to be
thermodynamically discriminated (15).
Distribution and Physiology of serotonin receptors
Human - In the central nervous system (CNS) 5-HT3 receptor is found in high densities in nuclei of the lower brainstem, area postrema and nucleus of the tractus solitarius. Lower densities of the receptor are found in the cerebral cortex and limbic areas, including the hippocampus. In the peripheral nervous system 5-HT3 receptors are located on pre- and postganglionic neurons of both sensory and enteric nervous systems (1). Nausea and vomiting are common adverse effects of therapeutic drugs. Such symptoms are more often due to CNS effects than to direct toxic effects on the gastrointestinal tract (GIT). Drugs may cross the blood-brain barrier and activate the chemoreceptor trigger zone in the brainstem, which contains cells that are responsive to cholinergic, dopaminergic and serotonergic stimulation. Selective serotonin (5-hydroxytryptamine; 5-HT) reuptake inhibitors (SSRIs) are effective and well tolerated in the treatment of major affective disorders, but their usefulness is sometimes limited by adverse effects, particularly gastrointestinal effects. SSRIs exert their beneficial effects in depressive syndromes by increasing brain serotonin levels. They also increase serotonin levels in other tissues, particularly the GIT, which contains 90% of the body's store of serotonin and large nos. of serotonin-responsive cells.
Increased serotonergic neurotransmission causes anorexia, nausea, vomiting and diarrhoea in other settings, such as carcinoid syndrome, so gastrointestinal adverse effects are not unexpected with drugs that increase tissue serotonin levels. SSRI-induced nausea and vomiting are probably due to effects on the GIT as well as on the CNS. There are complex interactions between serotonin receptor subtypes. Drugs antagonising one receptor subtype may act as agonists at another receptor. The pharmacotherapy of SSRI-induced nausea and vomiting requires an understanding of the actions and interactions of these receptors and their agonists/antagonists. The most effective drug for the treatment of SSRI-related adverse effects on the GIT is ondansetron, a serotonin 5-HT3 receptor antagonist that blocks the effects of serotonin in the brain and GIT. However, this drug has a high acquisition cost. Thus, the drug of choice may be cisapride which, although a weak 5-HT3 receptor antagonist, has the potential to reduce or abolish SSRI-induced nausea. Many patients with mild adverse effects will not require specific pharmacotherapy, as the nausea tends to abate with prolonged treatment with SSRIs because of gradual desensitization of 5-HT3 receptors (7).
5-Hydroxytryptamine (5-HT) has profound effects on cardiac rate and force in a variety of animal species, including humans. The main initial response to 5-HT is a short-lasting bradycardia, mediated via a Bezold-Jarisch-like reflex and initiated by stimulation of 5-HT,3 receptors present on cardiac vagal afferents (these are all forms of arrhythmias). Once this bradycardia is suppressed, 5-HT induces cardiac stimulation which, true to its chameleonic nature is mediated by different mechanisms and receptors in different species (9).
Although serotonin has been shown to play an important role in peripheral
pain mechanisms, the specific subtypes of serotonin receptors involved
in pain and hyperalgesia remain poorly understood. To date, no previous
study has attempted to deterimine the presence of any serotonin receptor
subtype in human dorsal root ganglia (suspected to be involved in pain
sensation). The presence of the GPCR type 5-HT1Da,
5-HT1Db, 5-HT1E, 5-HT2A and 5-HT7 receptor
subtype mRNA was detected in dorsal root ganglia from three of the four
subjects. 5-HT1A receptor subtype mRNA was detected in one of the four
subjects. No 5-HT2C receptor subtype mRNA could be detected. Findings
from this study may direct further efforts to determine the role of serotonin
receptors in the peripheral nervous system (13).
Regulation of pain
Serotonin is involved in regulating nociceptive input (pain sensation)
at variuous levels of the nervous system. 5-HT released from platelets,
mast cells or nerve endings activates primary afferent fibres and produces
pain per se. This process is mediated by 5-HT3 receptors since it can
be blocked by tropisetron (ICS 205-930). Torpisetron, however, has also
been shown to interfer with the binding of benzodiazepine (GABAA
receptor agonists). However, serotonin produces algetic or analgetic effects
depending on where it is realesed in the nervous system. In the spinal
cord and brain stem, serotonin exerts an inhibitory action on pain transmission
by increasing pain gating (5-HT1 and 2). Serotonin action in the peripheral
and central nervous system are mediated through different receptor subtypes
providing opportunities of the development of novel analgesic agents.
References
(1) Eglen, R. M.; Bonhaus, D. W. 5-Hydroxytryptamine (5-HT)3 receptors: molecular biology, pharmacology and therapeutic importance. Curr. Pharm. Des. (1996), 2(4), 367-374)
(2) Maricq, A. V.; Peterson, A. S.; Braker, A. J.; Myers, R. M. Kulius, D. Primary Structure and functional expression of 5HT3 receptor, a seretonin-gated ion channel. Science (1991) 254.432-437.
(3) Werner, Pia; Kawashima, Eric; Reid, John; Hussy, Nicolas; Lundstroem, Kenneth; Buell, Gary; Humbert, Yves; Jones, Kenneth A. Organization of the mouse 5-HT3 receptor gene and functional expression of two splice variants. Mol. Brain Res. (1994), 26(1-2), 233-41.
(4) Zeise, M. L.; Batsche, K.; Wang, R. Y. The 5-HT3 receptor agonist 2-methyl-5-HT reduces postsynaptic potentials in rat CA1 pyramidal neurons of the hippocampus in vitro. Brain Res. (1994), 651(1-2), 337-41.
(5) Van Hooft, J.A.; Vijverberg, H.P.M. Full and partial agonists induce distinct desensitized states of the 5-HT3 receptor. J. Recept. Signal Transduction Res. (1997), 17(1-3), 267-277.
(6) Glitsch, Maike; Wischmeyer, Erhard; Karschin, Andreas. Functional characterization of two 5-HT3 receptor splice variants isolated from a mouse hippocampal cell line Pfluegers Arch. (1996), 432(1), 134-143.
(7) Mcmanis, Philip G.; Talley, Nicholas J. Nausea and vomiting associated with selective serotonin reuptake inhibitors: incidence, mechanisms and management. CNS Drugs (1997), 8(5), 394-401.
(8) Localization of 5-HT3 receptors in the rat brain using [3H]LY278584. Gehlert, Donald R.; Gackenheimer, Susan L.; Wong, David T.; Robertson, David W. Brain Res. (1991), 553(1), 149-54.
(9) Saxena, Pramod R.; Villalon, Carlos M. 5-Hydroxytryptamine: a chameleon in the heart. Trends Pharmacol. Sci. (1991), 12(6), 223-7.
(10) Abi-Dargham, Anissa; Laruelle, Marc; Wong, David T.; Robertson, David W.; Weinberger, Daniel R.; Kleinman, Joel E. Pharmacological and regional characterization of [3H]LY278584 binding sites in human brain. J. Neurochem. (1993), 60(2), 730-7.
(11) Hamik, Anne; Peroutka, Stephen J. Differential interactions of traditional and novel antiemetics with dopamine D2 and 5-hydroxytryptamine3 receptors. Cancer Chemother. Pharmacol. (1989), 24(5), 307-10.
(12) Michel, Klaus; Sann, Holger; Schaaf, Cornelia; Schemann, Michael. Subpopulations of gastric myenteric neurons are differentially activated via distinct serotonin receptors: projection, neurochemical coding, and functional implications. J. Neurosci. (1997), 17(20), 8009-8017.
(13) Pierce, P. A.; Xie, G.-X.; Meuser, T.; Peroutka, S. J. 5-Hydroxytryptamine receptor subtype messenger RNAs in human dorsal root ganglia: a polymerase chain reaction study. Neuroscience (Oxford) (1997), 81(3), 813-819.
(14) Butler, A.; Elswood, C. J.; Burridge, J.; Ireland, S. J.; Bunce, K. T.; Kilpatrick, G. J.; Tyers, M. B. The pharmacological characterization of 5-HT3 receptors in three isolated preparations derived from guinea pig tissues. Br. J. Pharmacol. (1990), 101(3), 591-8.
(15) Borea, Pier Andrea; Dalpiaz, Alessandro; Gessi, Stefania; Gilli, Gastone. Thermodynamics of 5-HT3 receptor binding discriminates agonistic from antagonistic behavior. Eur. J. Pharmacol. (1996), 298(3), 329-34.
(16) Laguerre, Michel; Dubost, Jean Pierre; Kummer, Evelyne; Carpy, Alain. Molecular modeling of 5-HT3 receptor antagonists: geometrical, electronic and lipophilic features of the pharmacophore and 3D-QSAR study. Des. Discovery (1994), 11(3), 205-22, 3 plate.
(17) Schmidt, Anne W.; Peroutka, Stephen J. Quantitative molecular analysis predicts 5-hydroxytryptamine3 receptor binding affinity. Mol. Pharmacol. (1990), 38(4), 511-16.