Chromosome Organization: Nucleosomes
1. Introduction
Eukaryotic chromosomes are supramolecular complexes of DNA and protein. They are densely packed structures depending on the stage of the cell cycle. During cell division, or mitosis, the chromosome has its highest packaging with the usual four branches that are typically known as 'chromosome structure'. Two forms of chromatin have been described during the resting state of the cell. First, the heterochromatin, a densely packed form of the chromosomes, and second euchromatin, a less dens form with clearly discernible spherical particle, called nucleosomes, for every 300 base pair of DNA. Only in the form of euchromatin has gene expression been correlated. Its packing arrangements, protein-DNA interaction, and current models how chromosome structure is related to gene expression will be discussed here.
2. Histone proteins
The proteins that build the scaffold of the nucleosome are called histones. They form a family of five major classes of histone proteins called H1 (H5), H2A, H2B H3, and H4. The amino acid sequences of histones are highly conserved during evolution indicating their critical function for the chromosome organization and control of gene expression with the highest freqeuncy of mutations found in H1 (H5). This histone type has a special function in the nucleosomal complex at the nucleosome surface.
Histone proteins are basic proteins with a large proportion of positively charged amino acids, mainly Arg and Lys (9-30%). Histones can be posttranslationally modified through methylation, acetylation, and phosphorylation of specific Arg, His, Lys, Ser, and Thr residues. Acetylation of K and R residues at the N-terminal ends of H4 and H3 reduces the positive charges of histone proteins and destabilizes higher order nucleosome packing within the 300Å chromatin filament. The degree of modification varies with species, tissue, and stage of cell cycle. The highly conserved nature of the histone genes is therefore reduced through chemical modification and under enzymatic control of the cell. It is clear that these modifications must play a specific role in the control of DNA storage and expression. Why are histone proteins highly conserved? A possible answer is that mutations in histone genes critically affect posttranslational modification and hence chromosome organizatio, DNA transcription and replication.
3. Nucleosomes
Chromatin contains roughly equal numbers of molecules of histones H2A, H2B, H3, and H4, and no more than half that number of H1. Chromatin consists of ~ 100Å diameter particles, called nucleosomes, connected by thin strands of protein free DNA, so called linker DNA (electron microscopy). This has been shown by degradation of protein free DNA using nucleases. DNA in nucleosome particles, in contrast, is protected against micrococcal nuclease activity, because of its close interaction with histone proteins. The length of the protected DNA within each particle is about 200 base pairs. The core of each nucleosome consists of a histone octamer with a subunit stoichiometry of (H2A-H2B)-(H3-H4)-(H3-H4)-(H2A-H2B). A 146bp strand of DNA is wrapped around the histone octamer in 1.65 turns of a left-handed superhelix. Together, histonone octamer and 146bp of DNA constitute the nucleosome core particle. All core histones form a common fold of a central helix flanked on each side by a loop and a shorter helix (Voet&Voet, Fig. 33-7b). The H1 (linker histone) seals of the nucleosome and is likely to be responsible for control of gene expression (in the presence of some subtypes of H1, i.e., H5, DNA replication is inhibited).
The supramolecular nature of the core particle can be demonstrated in vitro. When purified DNA is mixed with equimolar amounts of histones, chromatin X-ray pattern can be seen similar to those of native nucleosomes. This means that the core particle self-assembles in vitro if the salt concentration is high and the histone concentration carefully controlled. Under physiological conditions, but still in vitro, histones tend to precipitate by interacting non-specifically with each other. To prevent this precipitation the cell provides an acidic chaperone protein that guides the core complex formation. This chaperone protein is called nucleoplasmin.
Fig. Nucleosome crystal structure at 2.8 angstrom resolution showing a disk-like shape
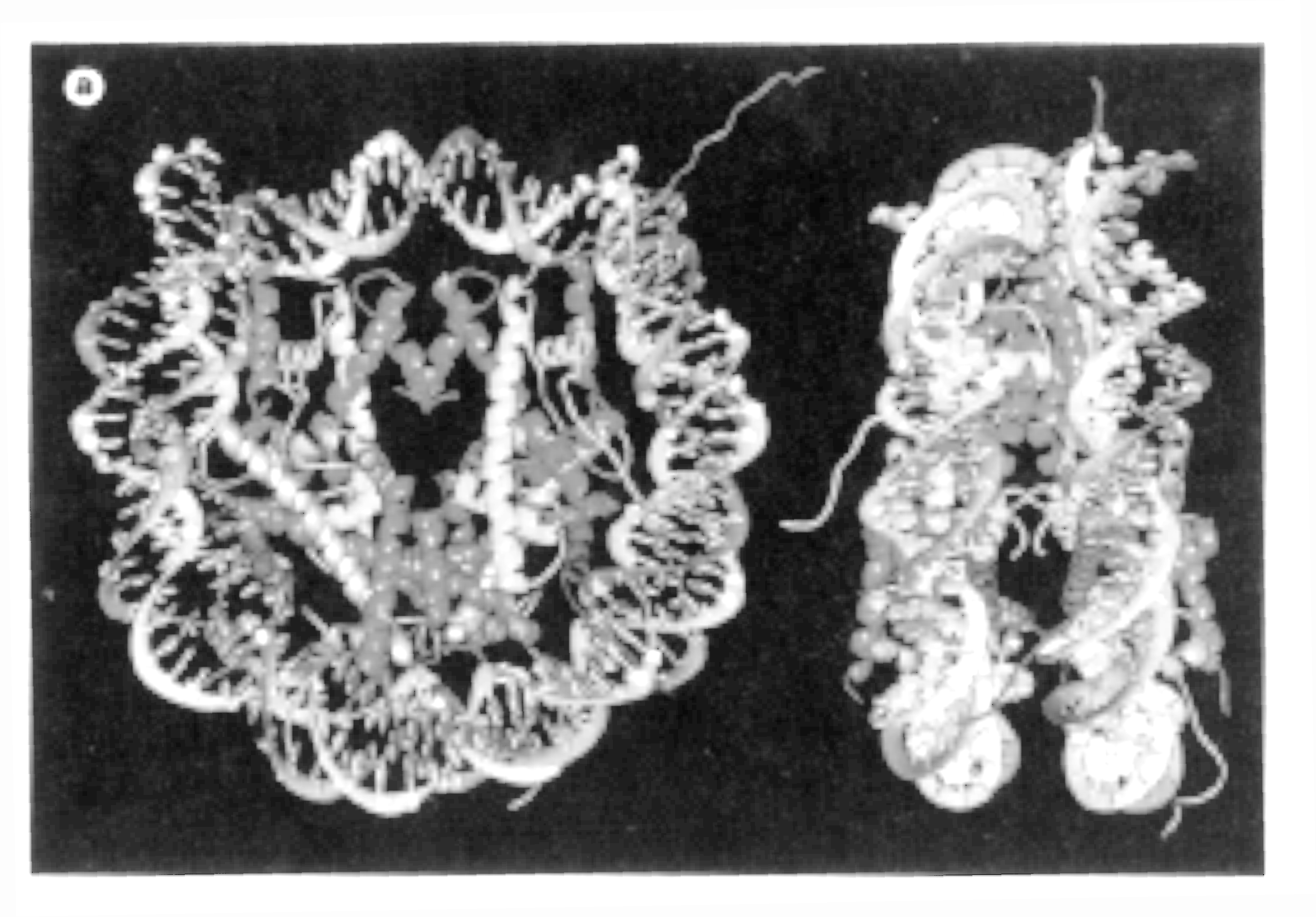
Note: the freely emerging unorderd structures are the N-terminal ends of H3 and H4 (from Luger, 1997)
The high resolution X-ray structure of the nucleosome core particle (histone
octamer containing H2A, H2B, H3, H4, and 146pp of DNA) has been solved
in 1997 using recombinant histone genes expressed in E.coli. Purified
and folded histone proteins assemble into a histone octamer and bind the
necessary core particle DNA fragment of 146 base pairs assuming that the
present structure represents the native nucleosome particle structure.
- histone monomers are defined by a three helix domain, called histone fold, with two unstructured tails- DNA enters and leaves the nucleosome at H3 contact sites (13bp at each entry site)
- each histone dimer binds about 30bp of DNA
- there are a total of 14 contact sites, each exhibiting a different curvature in the DNA double helix structure,
- contacts occur every 10.2bp with loops of histone fold (protein backbone) facing the minor groove through a total of 142 hydrogen bond interactions between histones and DNA
- phosphate backbone interacts with main chain atoms and interacting with Arg residues of histones providing a sequence independent mode of interaction.
4. Nucleosome-nucleosome packing and control of gene expression
The N-terminal ends contain large numbers of Lys and Arg residues which can be acetylated. Because these N-terminal ends provide inter-nucleosome contacts, acetylation modifies the degree of chromatin packing. Acetylation has been correlated with increased transcriptional activity. On the molecular level acetylation reduces the number of positive charges at the N-terminal ends which protrude from the core structure (see figure) and which interact with conserved negatively charged binding sites on neighboring nucleosomes (H2A-H2B dimer surface). Acetylation reduces high order structure of chromatin (stabilizes euchromatin) thus promoting gene expression activity.
Other evidence showing that histones are important for control of gene expression comes from similarities between proteins involved in transcription and histones. Recent evidence suggests that TBP-associated factors (TAF; for a discussion of TBP see section 3.2) contain sequences showing some similarity to histones of the nucleosome core particle (van Holde and Zlatanova, 1996). Not only has it been shown that these TAFs form histone core-like structures in the transcription initiation complex for RNA polymerase II, but individual subunits in heterotetramers formed from N-terminal fragments of Drosophila TAFs adopt the histone fold. The quaternary structure of the tetramer is similar to the H3-H4 tetramer in the nucleosome core. Based on the structural similarities between histone proteins and TAFs, it has been suggested that histone proteins might be involved in controlling transcription as do TAFs.
Fig. Structures of TAF (left) and H3-H4 dimers (right)
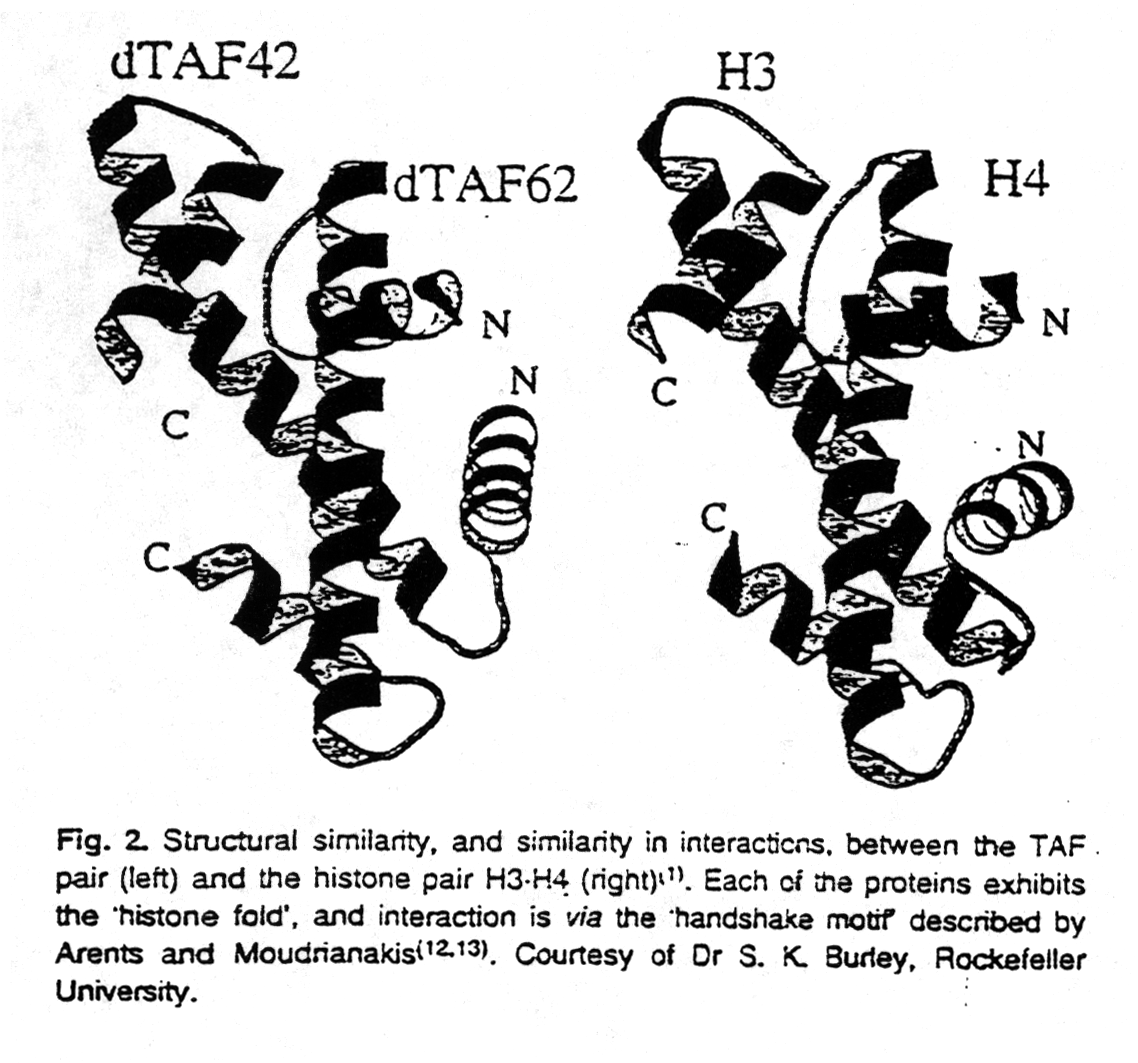
from van Holde and Zlatanova, 1996
Protein - DNA Interaction:
Transcription factors
1. General
Nucleic acids are the storage of genetic information and this information has to be accessible and inherited. The regulation of gene expression and the replication of the genome are central mechanisms to provide a 'translation' of the genetic blueprint into the machinery of life - the proteins. Nucleic acids need proteins for storage, replication, and transcription purposes. The specificity of the transcription and replication requires recognition on the molecular level between protein structures and nucleic acid structures. The genetic code has to be readable. The reading of the code is a conformationally specific interaction between amino acids and nucleic acids. Surface properties of the macromolecules involved are the essential key in the recognition process. Electrostatic interaction, hydrogen bonding capability and hydrophobic effects are of importance. Complementarity in surface profiles is the essential mechanism that provides the specificity. Here we discuss a few selected protein systems that recognize DNA double helical structures. The sequence of the base pairs in the double helix specifies the local conformation of the double helix - its ribose-phosphate backbone and the dimensions of the minor and major grooves of the helix.
Proteins that bind DNA and are involved in replication or transcription do so in a sequence specific way. Transcription factors are dimers when active, i.e., they bind to DNA upon dimerization and are inactive in the monomeric form. Dimerization is a regulatory mechanism of controlling transcription factor activity. There are 3 common features most DNA binding proteins have in common:
- the minor groove of B-DNA is 5Å wide, 8Å deep, and is generally too narrow to fit entire a -helices, but is recognized by b -sheet structures of TATA box binding proteins
- sequence specific DNA binding proteins generally do not disrupt
the base pairs of the DNA, but do distort backbone conformation
by bending the double helix
The helix-turn-helix motif is the common DNA recognition motif in prokaryotes (Voet&Voet, Fig. 29-22b). The motif resembles that of an EF hand described in calmodulin. The F-helix is the recognition helix and the side chains give the specificity of binding. Sometimes more than one protein compete for the same sequence. As examples serve bacteriophages l , P22, and 434, where repressors and activators affect transcription. They can recognize the same DNA fragment. The phage 434 cI repressor (Voet&Voet, Fig. 29-23) and cro (Voet&Voet, Fig. 29-24) have been shown in vitro to bind to the same 20bp DNA fragment. They have different binding interaction visualized by the two close, but not identical structures as determined by X-ray crystallography. They differ in their affinity for the same sequence, or DNA conformation, respectively through H-bonds, salt bridges and Van der Waals interactions. The relative concentrations of all proteins, therefore, determine which one is bound to the binding element most of the time, which in turn affects polymerase binding to the DNA determining if and in which direction transcription will occur. The ratio of all DNA binding proteins therefore determines the rate of transcription controlled by the DNA sequences in question.
3. Leucine-zipper (Eukaryotic transcription factors)
In some transcription factors the dimer binding site with the DNA forms a so called leucine zipper. This motif consists of two amphipathic helices, one from each subunit, interacting with each other resulting in a left handed coiled-coil super secondary structure (Voet&Voet, Fig. 33-56). The leucine zipper is a interdigitation of regularly spaced leucine residues in one helix with leucines from the adjacent helix. Mostly the helices involved in leucine zippers exhibit a heptad sequence (abcdefg) with residues a and d being hydrophobic and all others hydrophilic. Leucine zipper motifs can mediate either homo- or heterodimer formation. Note that the leucine zipper motif itself is not the DNA binding part of the helices.
Fig. Helical wheel representation of heptad sequence found in leucine zipper motif
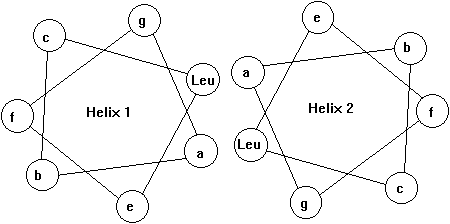
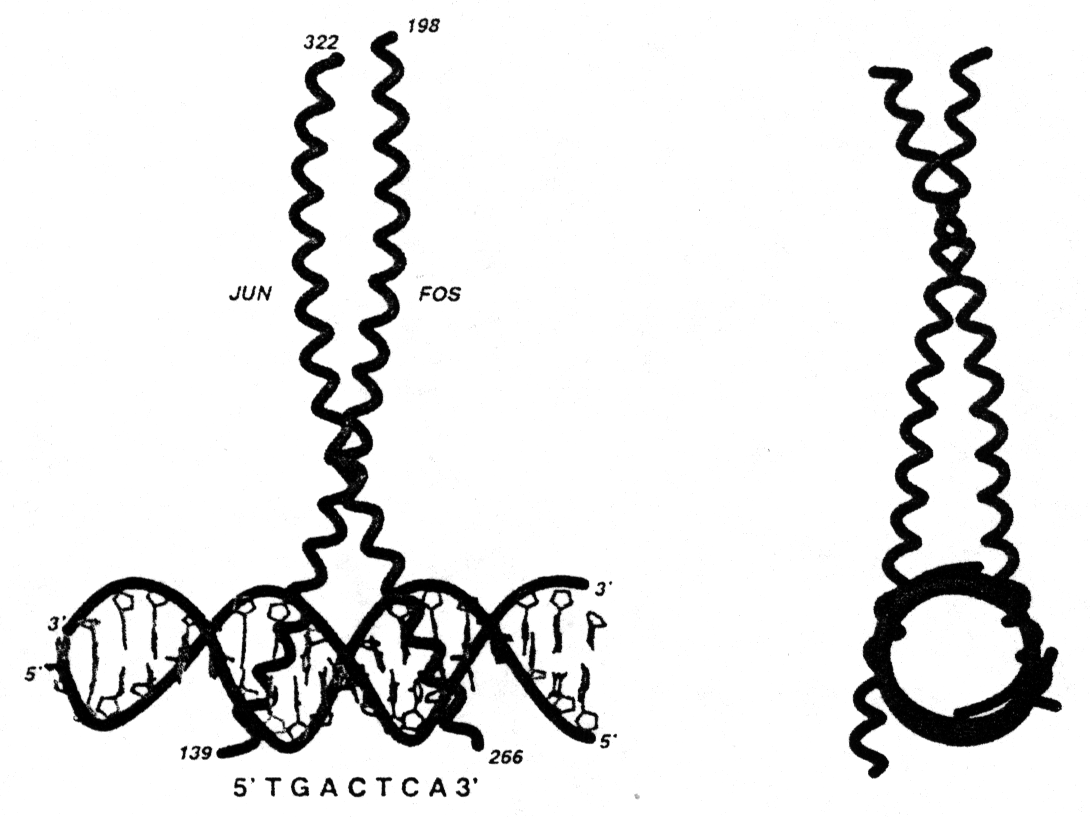
Fig. Structure of a leucine zipper - DNA complex (from Glover and Harrison, 1995)
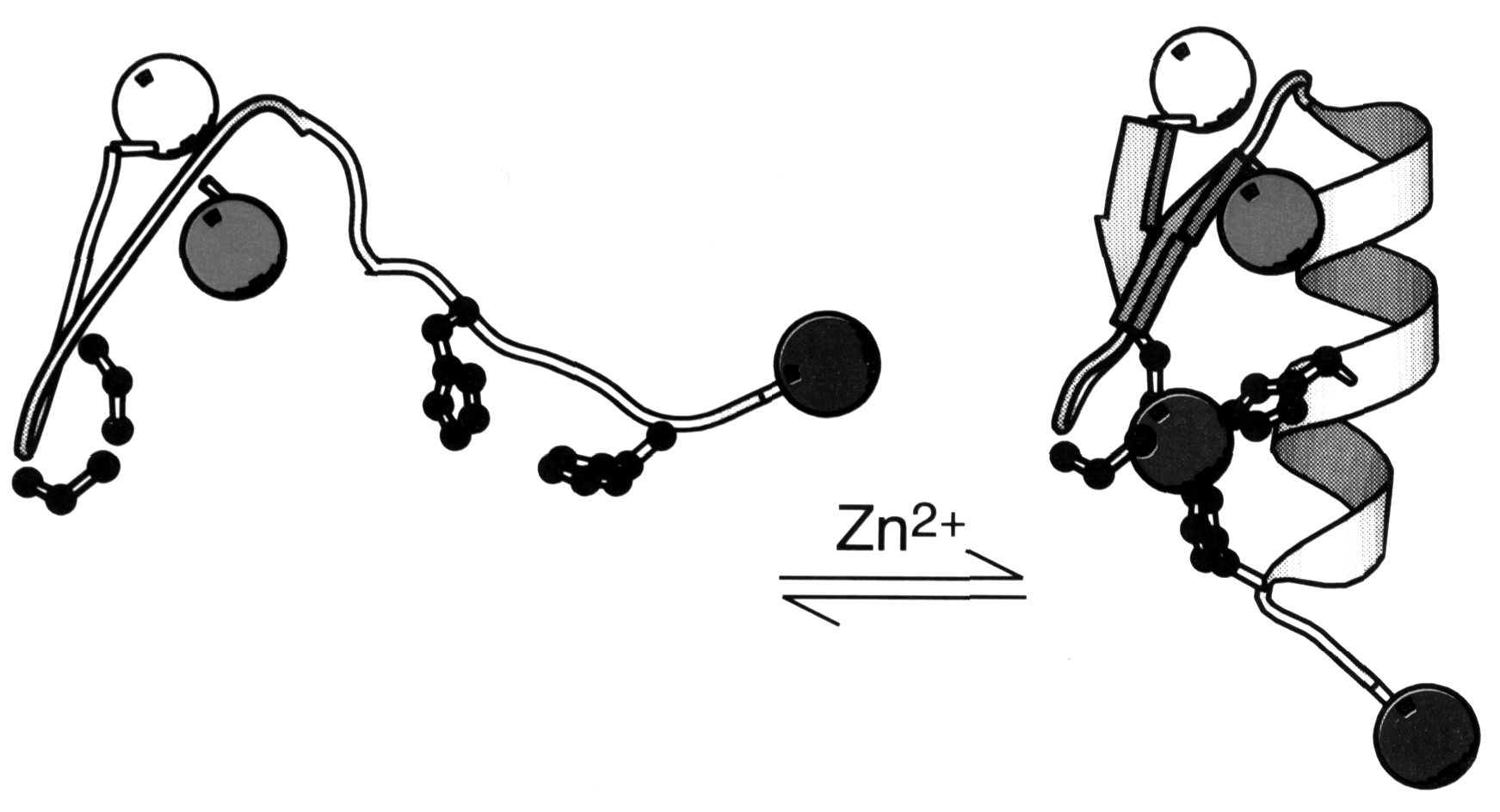
Some eukaryotic transcription factors showed a unique motif called a Zn-finger where a Zn++ ion is coordinated by 2 Cys and 2 His residues (Voet&Voet, Fig. 33-52). The transcription factor consists of a trimer with the stoichiometry bb 'a . The apparent effect of the Zn++ coordination is the stabilization of a small loop structure instead of hydrophobic core residues. Each Zn-finger interacts in a conformationally identical manner with successive triple base pair segments in the major groove of the double helix. The protein-DNA interaction is determined by two factors: (i) H-bonding interaction between a -helix and DNA segment, mostly between Arg residues and Guanine bases. (ii) H-bonding interaction with the DNA phosphate backbone, mostly with Arg and His. An alternative Zn-finger motif chelates the Zn++ with 6 Cys (Voet&Voet, Fig. 33-55). Note that in all cases the Zn++ does not itself participate in binding interaction.
Fig. Zn-finger peptide undergoes folding transition upon metal binding
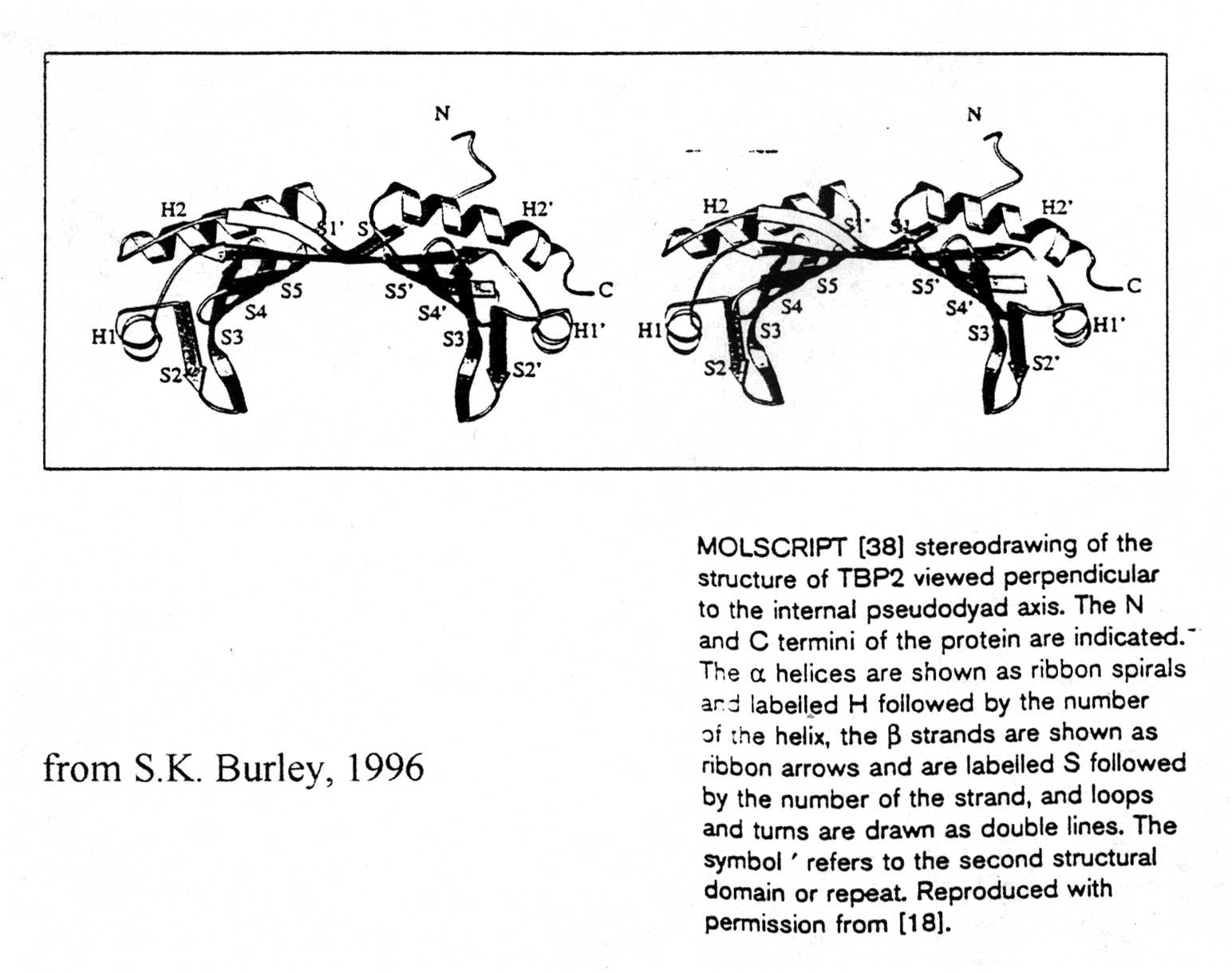
TATA box binding proteins were first identified as a component of the
class II initiation factor TFIID. They participate in transcription by
all three nuclear RNA polymerases (S.K. Burley, 1996) acting as subunit
in each of them. The structure of TBP was solved at 2.1Å resolution
showing two a /b
structural domains of 89-90 amino acids. The C-terminal or core region
binds with high affinity to the TATA consensus sequence (TATAa/tAa/t)
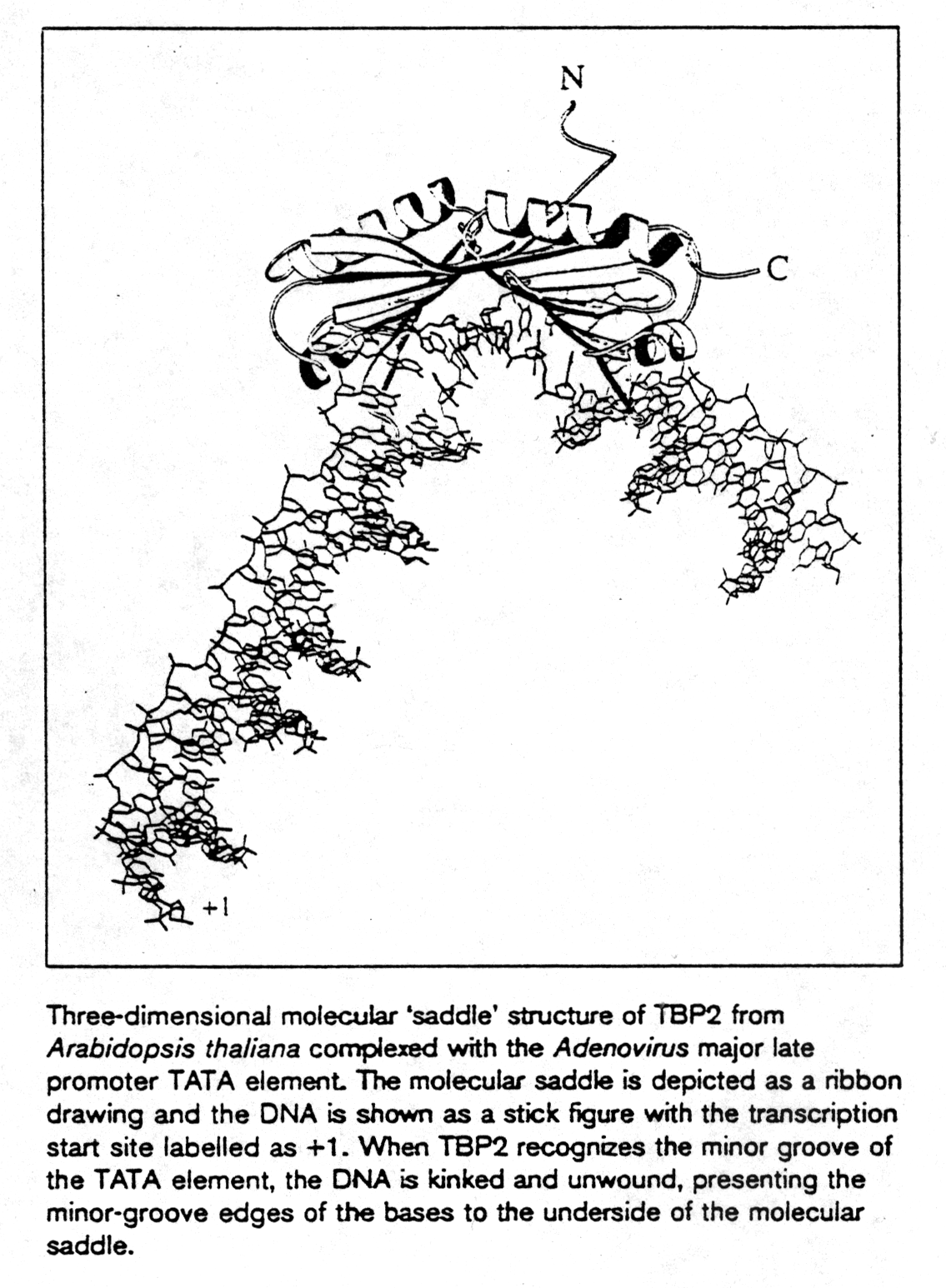
recognizing minor groove determinants and promoting DNA bending as shown.
TBP resembles a molecular saddle with approximate dimensions 32Åx45Åx60Å.
The binding side is lined with the central 8 strands of the 10-stranded
anti-parallel b -sheet. The upper surface contains
four a -helices and binds to various components
of the transcription machinery.