Drug-DNA interaction
DNA as carrier of genetic information is a major target for drug interaction because of the ability to interfere with transcription (gene expression and protein synthesis) and DNA replication, a major step in cell growth and division. The latter is central for tumorigenesis and pathogenesis.
There are three principally different ways of drug-binding. First, through control of transcription factors and polymerases. Here, the drugs interact with the proteins that bind to DNA. Second, through RNA binding to DNA double helices to form nucleic acid triple helical structures or RNA hybridization (sequence specific binding) to exposed DNA single strand regions forming DNA-RNA hybrids that may interfere with transcriptional activity. Third, small aromatic ligand molecules that bind to DNA double helical structures by (i) intercalating between stacked base pairs thereby distorting the DNA backbone conformation and interfering with DNA-protein interaction or (ii) the minor groove binders. The latter cause little distortion of the DNA backbone. Both work through non covalent interaction.
The small ligand drug approach offers a simple solution. The synthesis
and screening of synthetic compounds that do not exist in nature, work
much like pharmacological ligand for cell surface receptors in excitable
tissue, and appear to be more readily delivered to cellular targets than
large RNA or protein ligands. The lack of sequence specificity for intercalating
molecules, however, does not allow to target specific genes, but rather
certain cellular states or physiological and pathological conditions,
like rapid cell growth and division that can be selectively suppressed
as compared to non growing or slowly growing healthy tissue.
Modeling DNA-ligand interaction of intercalating ligands
The following properties have been identified as important for the successful modeling of ligand-DNA interaction:
- degrees of freedom
- role of base pair sequence
- counter ion effects
- role of solvent ligand-receptor binding
- degrees of freedom
This problem is analogous to that of protein ligand interaction. The major
requirement for intercalating agents is the planar aromatic ring structure.
This structure fits between to adjacent base pair planes and can have
some, although much restricted, rotational freedom within the plane of
the ring. The ligand itself may have flexibility of structural parts outside
the DNA binding site and may contain more than one intercalating sidechain:
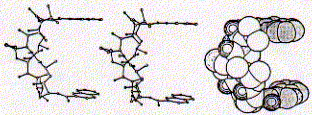
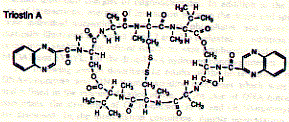
The structure of the antibiotic triostin A shows the presence of two quinoxaline (groups to the right; double aromatic rings) units linked through a cyclic peptide structure (center left) which is stabilized at its center by a cystein pair (disulfhydril covalent bond).
Fig. Chemical structure of triostatin A
The space filled side view indicates how the two quinoxaline rings are positioned by the linker peptide in co-planar fashion suitable for intercalating with DNA base pairs. As a rule, the more intercalating sidechains are linked within a single ligand structure, the stronger the expected binding affinity.
Triostatin A belongs to a family of antibiotics which are characterized by cross-linked octapeptide rings bearing two quinoxaline chromophores. Since the spacing between the chromophores is 3.5A, the intercalation process sandwiches two base pairs between the two quinoxalines. This phenomenon is called bis-intercalation and has first been described for echinomycin by showing that bis-intercalating drugs cause twice the DNA helix extension and unwinding seen as compared to single intercalating molecule like ethidium. The latter is a chromophore which is activated by UV light and is used by molecule biologists to label nucleic acids in gel electrophoresis or ion gradient centrifugation.
- role of base pair sequence
experimental evidence suggests that base pair sequence does not play a
large role on the specific mature of most intercalating complexes. As
the structure of triostatin A suggests, however, the linker peptide structure
may well promote specific interaction with the DNA surface. The major
group specific readout sequence of H-bond donor and acceptor could be
involved in triostatin A binding. The figure graphically shows the direct
readout of the DNA base sequence on a double helical structure.
Fig. Overlap of AT and GC base pairs
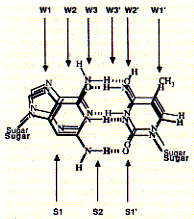
Note: readout sequence of minor (S) and major groove (W)
side as they are available for protein interaction.
The following characteristics of non covalent bond formation are associated
with the binding sites indicated above:
| binding site | GC base pair | AT base pair |
| W1 | H-bond acceptor | H-bond acceptor |
| W2 | blank | blank |
| W3 | H-bond acceptor | H-bond donor |
| W3' | blank | blank |
| W2' | H-bond donor | H-bond acceptor |
| W1' | C-H weak hydrophobic | CH3, strong hydrophobic |
While the interaction on the major groove side is distinct for the direction of the base pair (e.g. AT vs TA), there is no directionality at the minor groove side.
The molecular basis of specific recognition between echinomycin and DNA is due to the hydrogen bonding between the ligand alanine carbonyl groups and the 2-amino group of guanine. This is consistent with the observation that the preferred binding site is the sequence CG
- counter ion effect
DNA is a negatively charged polyanion attracting counter ions, positively
charged Na+, or Ca++ and Mg++ ions as well as basic residues of proteins.
The presence of small counter ion affect drug binding, since the counter
ions can screen and shield the negative backbone surface allowing non
electrolytes as well as positively charged ligand to interact more strongly
with the DNA target. High ionic strength, however, reduces non covalent
interaction mediated by hydrogen bonds and electrostatic interactions.
- role of solvent ligand-receptor binding
There are three general classes of interactions that must be considered
in solvated ligand-receptor binding
(a) ligand solvent interaction (e.g. hydration shell), (b) receptor solvent
interaction, and (c) ligand-DNA complex with solvent interaction. The
three classes basically describe the sequence of events of free ligand
interacting with its receptor and the change in overall solvent interaction
before and after binding. We have seen that the hydrophobic effect is
completely described by this system and the contribution of the entropy
of free bulk water is the major driving force of hydrophobic ligand receptor
interaction. This type of interaction is found in intercalating substrates
because the hydrophobic, aromatic sidechains interactive favorably with
the aromatic environment of the base pair stacking. The total amount of
surface bound water is reduced in the after complex formation.
- rational for drug design
When a compound intercalates into nucleic acids, there are changes
which occur in both the DNA and the compound during complex formation
that can be used to study the ligand DNA interaction. The binding is of
course an equilibrium process because no covalent bond formation is involved.
The binding constant can be determined by measuring the free and DNA bound
form of the ligand. Since many of the intercalating substrates are aromatic
chromophores, this can be done spectroscopically. Also, DNA double helix
structures are found to be more stable with intercalating agents present
and show a reduced heat denaturation. Correlating these biophysical parameters
with cytotoxicity is used to support the antitumor activity of these drugs
as based on their ability to intercalate in DNA double helical structures.
Improvement of anticancer drugs based on intercalating activity is not
only focussed on DNA-ligand interaction, but also on tissue distribution
and toxic side effects on the heart (cardiac toxicity) due to redox reduction
of the aromatic rings and subsequent free radical formation. Free radical
species are thought to induce destructive cellular events such as enzyme
inactivation, DNA strand cleavage and membrane lipid peroxidation.
Modeling DNA-ligand interaction of minor groove binders
Hairpin minor grove binding molecules have been identified and synthesized that bind to GC reach nucleotide sequences. Hairpin polyamides are linked systems that exploit a set of simple recognition rules for DNA base pairs through specific orientation of imidazole (Im) and pyrrole (Py) rings. The hairpin polyamides originated from the discovery of the three-ring Im-Py-Py molecule that bound to minor groove DNA as an antiparallel side by side dimer.
Fig. Structure of hairpin ligand (right) on DNA minor groove (left)

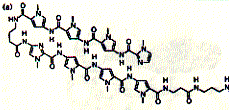
from JB Chairs, Current Opinion in Structural Biology, 1998,
8:314-320
The compound was found to recognize GC base pairs. Solid phase synthesis of polyamides of variable length has produced efficient ligands, e.g. the eight ring hairpin polyamide ImPyPyPy-g-ImPyPyPy-b-Dp (Dp dimethylamino propylamide) shown in the figure above. This small synthetic molecule has an binding constant in the order of 0.03nM.
The optimal goal of polyamide ligand design has been reached with finding structures able to recognize DNA sequences of specific genes. The structure shown above inhibits the expression of 5S RNA in fibroblast cells (skin cancer cells) by interfering with the transcription factor IIIA-binding site.
A new strategy of rational drug design exploits the combination of polyamides with bis-intercalating structures. WP631 is a dimeric analog of the clinically proven anthracycline antibiotic daunorobuicin.
Fig. Structure of WP631
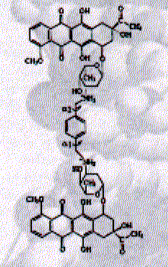
from JB Chairs, Current Opinion in Structural Biology, 1998,
8:314-320
This new synthetic compound shows an affinity of 10pM and also showed
to be resistant against multidrug resistance mechanisms often encountered
in antitumor therapy. Multidrug resistance is a phenomenon where small
aromatic compounds are efficiently expelled from the cell by cell membrane
transport proteins commonly referred to as ABC transporters (or ATP Binding
Cassette proteins).
Drugs that form covalent bonds with DNA targets
Drugs that interfere with DNA function by chemically modifying specific nucleotides are Mitomycin C, Cisplatin, and Anthramycin.
Mitomycin C is a well characterized antitumor antibiotic which forms a covalent interaction with DNA after reductive activation. The activated antibiotic forms a cross-linking structure between guanine bases on adjacent strands of DNA thereby inhibiting single strand formation (this is essential for mRNA transcription and DNA replication).
Anthramycin is an antitumor antibiotic which bind covalently to N-2 of guanine located in the minor groove of DNA. Anthramycin has a preference of purine-G-purine sequences (purines are adenine and guanine) with bonding to the middle G.
Cisplatin is a transition metal complex cis-diamine-dichloro-platinum
and clinically used as anticancer drug.
 The effect of
the drug is due to the ability to platinate the N-7 of guanine on the
major groove site of DNA double helix. This chemical modification of platinum
atom cross-links two adjacent guanines on the same DNA strand interfering
with the mobility of DNA polymerases.
The effect of
the drug is due to the ability to platinate the N-7 of guanine on the
major groove site of DNA double helix. This chemical modification of platinum
atom cross-links two adjacent guanines on the same DNA strand interfering
with the mobility of DNA polymerases.