Enzyme Catalysis - Serine Proteases
1. The role of proteases in the cell
Proteases are proteins that cleave, or cut, or degrade other proteins
by hydrolyzing peptide bonds. This may sound like an uninteresting
topic, but protein degradation plays a major role in cellular processes.
Proteases are involved in cellular control mechanisms:
- removal of old and unused proteins. This is an essential part of the turnover pathway of all proteins in the cells and affects cell growth.- degradation of proteins and peptides for nutritional purposes
- defense mechanisms against intrusive proteins and peptides
- control of protein activity
2. Classification of proteases
The proteases are globular, water soluble proteins that function as enzymes.
They catalyze the hydrolyses of peptide bonds in proteins. Being enzymes,
proteases can be characterized by their substrate affinity and
the catalytic rate of the reaction. Using proteases to study the
effects of single amino acid substitutions (mutations) on catalytic rate
and substrate affinity demonstrated that these two properties are linked
and that this linkage can be explained by analyzing the conformation of
the catalytic or active site of the enzyme. This analysis showed that
four major functional groups are found in the catalytic site of proteases
and based on this functional groups, 4 families of proteases have been
defined:
- Serine proteases- Cystein proteases
- Aspartate proteases
- Metallo proteases
This classification uses the functional group within the enzyme, and does
not relate to the substrate specificity of the proteases themselves.
The name of the protease family refers to an amino acid (e.g. aspartate)
or a metal as co-enzyme at the active site of the enzyme.
3. Enzyme catalysis - Michaelis-Menten Kinetic
In 1913 Michaelis and Menten described the reaction rate and specificity for a simple one-site reaction. Here is a simplified representation of their theory. Michaelis and Menten divided the process of the conversion of a substrate S into a product P into two steps as shown:
Km
kcat
E + S <=> ES <=> E + P
The first reaction step describes the binding of the substrate to the enzyme (catalyst) and the constant Km corresponds to the dissociation constant of the equilibrium under conditions where the product formation is very slow compared to the dissociation process of the substrate. Km equals the substrate concentration at half maximal reaction rate Vmax/2. In this case Km is a good apporoximation for the dissociation constant and thus describes the affinity of the substrate for the enzyme. For more complex reactions the constant reflects the dissociation equilibrium of all substrates bound to the enzyme.
The second reaction step describes the catalytic rate or the rate of product formation and referred to as the turnover number kcat. The turnover rate is defined as the maximal number of product P per active site per unit time.
The Michaelis-Menten kinetic is valid only under saturation conditions, i.e., when the concentration of substrate S is much larger than the enzyme concentration. The maximal reaction rate Vmax describes a steady-state equilibrium of the reaction catalyzed by the enzyme. The steady-state equilibrium is an important concept in biochemistry because many enzyme catalyzed reactions run at saturation and the product often is removed from the reaction site so as to render the reaction irreversible.
The saturation conditions of an enzymatic catalysis, where the substrate concentration exceeds the enzyme concentration, for all practical purposes, means that the Michaelis-Menten kinetic is valid only for initial rates because with time the substrate gets depleted and we no longer deal with an excess of substrate over enzyme. In addition, product P can affect the reaction, and this is indeed a major mechanism in the control of biochemical reactions. The product can often bind to the enzyme and at a site other than the active site influence its activity. This is called feed-back control, which can be positive or negative feed back, stimulate or inhibit the formation of P.
To complete this brief description of enzyme kinetics, we define the
substrate specificity. The ratio kcat / Km
defines a measure of the catalytic efficiency of an enzyme-substrate pair.
It refers to the properties and reactions of free enzyme and free substrate.
The theoretical limit for kcat / Km is set by the
rate constant of the initial complex formation (ES) and cannot be faster
than the diffusion controlled interaction of substrate and enzyme. The
specificity of an enzyme is therefore a measure of the specificity of
an enzyme for competing substrates or of competing enzymes for a single
substrate. Note that specificity here should not be confused with the
use of the term 'specificity' instead of 'affinity', as in 'specificity
pocket' discussed below.
4. Activation energy
The rate of an enzymatic reaction is strictly dependent on the activation
energy of the catalyzed reaction. Enzymes catalyze chemical reactions
by lowering the activation energy of the reaction. This is achieved
by either the binding of two substrates in an orientation optimal for
the reaction to occur, or by using differential binding energies for transition
state of S* as compared to the initial bound state of S (binds S* more
tightly than S).
5. Chymotrypsin super family of proteases
Chymotrypsin, trypsin, and elastase are three members of the super family
of chymotrypsin proteases. They all are serine proteases because the amino
acid residue at the catalytic site responsible for the transition state
stabilization is a serine. These three proteins have different overall
structures but identical active site conformations. The active site of
a serine protease can be divided into four essential structural features
required for the catalytic action of serine proteases:
| Structural element of active site | Function |
| The main chain substrate binding | unspecific binding of polypeptide segment |
| Specificity pocket | semi-specific binding of side chains, sequence specificity |
| Oxyanion whole | stabilizes S* over S in enzyme |
| The catalytic triad | forms tetrahedral intermediate (transition state; stabilizes S* over S); hydrolyzes peptide bond |
6. The reaction mechanism of chymotrypsin
The protease chymotrypsin is synthesized as an inactive precursor
protein, called Chymo-trypsinogen, consisting of 245 amino acids.
The enzyme is activated by the proteolytic removal of two di-peptides
at positions 14-15 and 147-148. The active protein consists of three polypeptide
chains covalently linked through S-S bonds, and non-covalently through
hydrogen bond formation and non-polar interactions. Therefore, the native
complex, although cleaved into three polypeptides, does not fall apart.
The protein consists of 2 domains that are approximately equal, containing
~120 amino acids each and are build from an anti-parallel b-barrel.
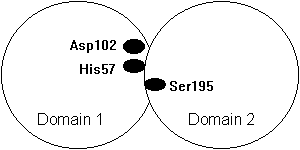
Fig. Domain structure of a chymotrypsin type serine protease with the catalytic triad at the domain interface (Asp102 and His57 in domain 1 and Ser195 in domain 2).
2. covalent bond formation between -C=O and S195 hydroxyl (nucleophilic attack), tetrahedral intermediate as transition state, oxyanion hole stabilization, H57 binds the -released H+ from S195 (ES*)
3. acyl-enzyme intermediate and peptide bond hydrolysis, peptide with new N-term released from enzyme taking -H from H57 (originally S195; see step 2) (EP*)
4. a water molecule placed next to H57 and acyl-enzyme intermediate at S195 (EP*)
5. the water molecule initiates a nucleophilic attack and undergoes a reaction with one -H attaching to H57 and -OH to acyl-enzyme intermediate forming again a tetrahedral transition state of the new C-terminal peptide fragment (EP*)
6. peptide released while H57 donates -H (from water molecule) to
S195 (E+P)
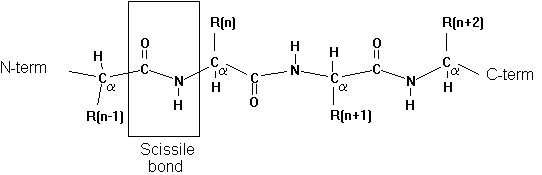
7. Specificity pocket
Proteases have preferential cleavage sites in the sequence of
a protein substrate. The specificity pocket provides a small binding pocket
consisting of 3 amino acid residues that determine the local polarity
and electrostatic potential profile for the interaction of residue n-1
on the substrate on the N-terminal side of the scissile bond. For the
chymotrypsin family of serine proteases we find the following sequence
specificities:
Table Specificity pocket substrate interaction for the Chymotrypsin Superfamily
| serine protease | chymotrypsin | trypsin | elastase |
| Rn-1 | bulky, aromatic | + charged | small, non-charged |
The comparison of the two families of serine proteases tells us two different
things. First, it has been reasoned to be an example of convergent
evolution, where the formation of a catalytic site has evolved twice,
with each protease family exhibiting a different overall 3-D structure.
Second, the differences in the 3-D structure gives us an idea of the different
cellular locations of the corresponding protein families: the catalytic
site of an enzyme is conserved over evolutionary time, while the overall
structure is conserved to provide structural stability for optimal activity
of the protein in any given environment. We need only understand that
very different sequences can provide similar 3-D structures because water
solubility depends only on the distribution of hydrophilic and hydrophobic
residues, but not on other chemical properties. Overall structural features
thus reflect the location of the protein, if it is located intra-cellular,
extra-cellular, if it is cell membrane protein, or if it is resistant
to temperature changes or sensitive to proton or calcium concentrations.
8. Protease Inhibitors
Cellular control of proteases is carried out by protease inhibitors.
These are small peptides or proteins that can bind to the active site
of the protease (competitive inhibitor) but which are not hydrolyzed,
thereby blocking the access for substrate proteins. One example of a protease
inhibitor is the bovine pancreatic trypsin inhibitor (BPTI), a
small protein of 58 amino acids. Its structure has been determined by
X-ray crystallography and the protein has been widely used for folding
studies of proteins (see section 2.7). BPTI binds to trypsin through hydrogen
bonding forming a tightly packed interface between inhibitor and enzyme.
The Michaelis-Menten constant of BPTI binding Km = 10-13M.
The lysine at position 15 binds to the specificity pocket followed by
an alanine. The reaction is blocked at the formation of the transition
state intermediate.
Allostery and Cooperativity - Hemoglobin
1. Introduction
Most enzymes are protein complexes. Single protein enzymes, such as serine proteases, are rare. The discussion of the structure-function relationship of the oxygen binding myoglobin and hemoglobin provides an insight to the advantage of polypeptide association into larger functional complexes. Protein complexes provide structural and functional variabilities through combinatorial effects of subunits that cannot be achieved by single polypeptides (of course they could be large, multi-domain proteins, where each domain adopts the function of a subunit). The single most important functional aspect, however, affects the regulation of the protein complex. Oligomerization enables cooperativity between multiple binding sites, a property referred to as allostery or allosteric regulation of enzymes (yet again, single polypeptide, multi-domain proteins show allosteric behavior).
Protein complexes are often referred to as oligomeric proteins. Oligomeric proteins are composed of subunits, with the subunits being individual polypeptide chains. Protein complexes have defined quaternary structural symmetries, if they are homo-oligomeric protein complexes where all subunits are identical. The quaternary symmetry are pseudo-symmetries if the complex is a hetero-oligomer. Homo- and hetero-oligomeric compositions allow for an additional regulatory hierarchy at the cellular, tissue, or organism level, where different combinations of subunits of heteromeric complexes, due to cell specific gene expression, often show minute, yet important differences in enzyme activity.
Two protein systems will be discussed in detail to elucidate the structure-function relationship in oligomeric proteins. The first system is the globin protein family. Members of this family are the myoglobin, a single polypeptide enzyme, and hemoglobin, a tetrameric protein complex with a2b2 stoichiometry (where the Greek letters denote subunit types, not secondary structure). Both globins are oxygen binding proteins. Myoglobin has one binding site, whereas hemoglobin has four binding sites, one per subunit. Myoglobin and hemoglobin differ in their regulatory capacity of binding O2. The four binding sites in hemoglobin show positive cooperative binding, meaning that after the first binding site is occupied by an O2 molecule, the 3 remaining sites exhibit a largely increased affinity for O2 molecules.
The second protein system is the nicotinic acetylcholine receptor,ornAChR, in post-synaptic membranes of skeletal muscle and neurons (see section 2.3). This receptor is a heteromeric protein complex of five integral membrane proteins with a subunit stoichiometry of a2bgd. The five subunits are arranged in a circular fashion around a central hole that provides an ion pathway across the cell membrane. The pentameric complex has a five fold pseudo-symmetry because its subunits are not identical. The receptor complex binds two molecules of acetylcholine. Acetylcholine binding induces the opening of the channel thereby transmitting a chemical signal (neurotransmitter binding) into an electrical signal (ion flux). The coupling of the ligand binding site and the channel structure (active site) is an allosteric effect because the binding of the ligand causes a structural change at a distant place on the receptor unit.
Both oligomeric protein systems, although they have completely different
functions, use a similar mechanism that makes a hemoglobin a cooperative
(crosstalk between identical units) unit and the nAChR an allosteric ion
channel (crosstalk between different units). The distance between two
heme binding sites in hemoglobin is about 2.5nm and the distance between
the acetylcholine binding site and the channel gate, the structure in
the complex that opens and closes the ion pathway across the membrane,
is 2.5nm as well.
2. Structure of Myoglobin and Hemoglobin
Myoglobin is a small globular protein that binds O2 in muscle tissue. Its 'enzyme' activity is not the catalysis of a reaction, but to increase the water solubility of oxygen and to facilitate its transport to the muscle cell. Its structure was the first to be solved in its entirety by X-ray crystallography (Kendall et al., 1961). It contains 153 amino acids and is folded into 8 a-helices, labeled A through H, connected through short loop structures. The a-helix motif of myoglobin is also called the globin fold. The O2 binding site consists of a protoporphyrin IX ring with a centrally coordinated Fe(II) forming the heme group which is non-covalently embedded in the globin fold between helices E and F. The central Fe(II) coordinates with the four N in the protoporphyrin IX structure. The fifth coordination site is occupied by His93 amine (helix F). The sixth coordination place is free and accessible for an O2 molecule. Because of its asymmetric coordination state in the oxygen free state, the Fe is slightly positioned off the protoporphyrin ring plane. The binding of an oxygen molecule pulls the Fe(II) into the plane of the heme. The His93 is pulled along with the Fe(II) and as a consequence displaces the F-helix at one end by about 0.1nm.
Comparing the amino acid sequences of related globins for the helix-helix contacts shows no similarity. Obviously different amino acids can be replaced without disrupting the over all globin fold. The heme-pocket, however, is an evolutionary conserved structure. Mutations in the globin fold that do not affect the conformation of the heme-pocket are accepted, even if they might slightly affect relative positions between helices or loop structures.
Hemoglobin is a tetrameric protein complex composed of two alpha and two beta subunits with an overall subunit stoichiometry of a2b2. The hemoglobin tetramer is a spheroidal protein complex with the dimension 6.4 x 5.5 x 5.0 nm. The tetramer has a pseudo-D2 symmetry and the association between subunits is stabilized by hydrophobic interactions as well as hydrogen bonds. The overall sequences and structures of the hemoglobin subunits are homologous to myoglobin. Their arrangement into a tetramer, built from the association of two ab dimers has important consequences with respect to oxygen binding affinity and regulation. Hemoglobin contains 4 heme groups, all of which are bound to each individual subunits as shown for myoglobin. The binding of the first O2 to any of the hemes induces a shift in the corresponding F-helix. This structural change within a subunit is sensed by the neighboring subunits in a way that the binding of O2 to their hemes is favored as compared to the initial conformational situation. Thus, the binding of a first O2 induces a decrease in the binding energy for subsequent O2 binding in the other three subunits.
This mechanism is called cooperative effect.
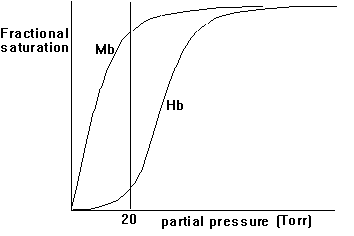
3. Hill coefficient - a measure of cooperativity
Both myoglobin and hemoglobin bind O2 and transport it to their target. Why is one type of globin not enough? Looking at the physiology of transporting oxygen to our muscle cells and kinetically analyzing the effects of cooperativity and non-cooperativity of O2 binding, we can observe a physiological system that provides both a high efficiency and tight control over O2 transport to the electron transport systems in muscle cells that convert the high energy content of O2 bond electrons upon oxidation into metabolic energy. To understand cooperativity of O2 binding we have to define the fractional saturation, YO2, of globin with O2 molecules. The fractional saturation can experimentally be determined by measuring the partial oxygen pressure, pO2, in solution. The fractional saturation and the partial oxygen pressure are related as shown in this equation:
p(O2)n
Y(O2) = -----------------
p(50)n +p(O2)n
p(50) is defined as partial pressure of O2 to upon 50% occupation
of all heme binding sites by ligands. The cooperativity factor is indicated
by the factor n; for hemoglobin n = 3; for myoglobin n = 1.
Fig. Fractional saturation of Myoglobin (Mb) and Hemoglobin (Hb)
4. Mechanism of cooperativity of O2 binding
The structural changes in Hb subunits can be analyzed and a kinetic model for those structural changes has been described. In this model, the quaternary structure of hemoglobin with no molecular oxygen bound, deoxyHb, is designated as T-state and the fully oxygenated form, oxyHb, as R-state. Similarly the subunit forms are designated t and r states. It is important to understand that the occupancy state of one heme cannot be sensed by an other heme by direct electronic interaction. They are too far away (>2.5nm). The interaction is a mechanical one, transmitted through the conformational change induced by the binding of one O2 to a heme and the consequent displacement of the F-Helix. This helix displacement brings upon a change in the quaternary structure such that the dimer (a1b1) rotates 15° relative to the a2b2 dimer. The dimers a1-b1 and a2-b2 relative subunit orientation is unaffected.
Fig. Quaternary conformational change in hemoglobin (Fermi, p.169)
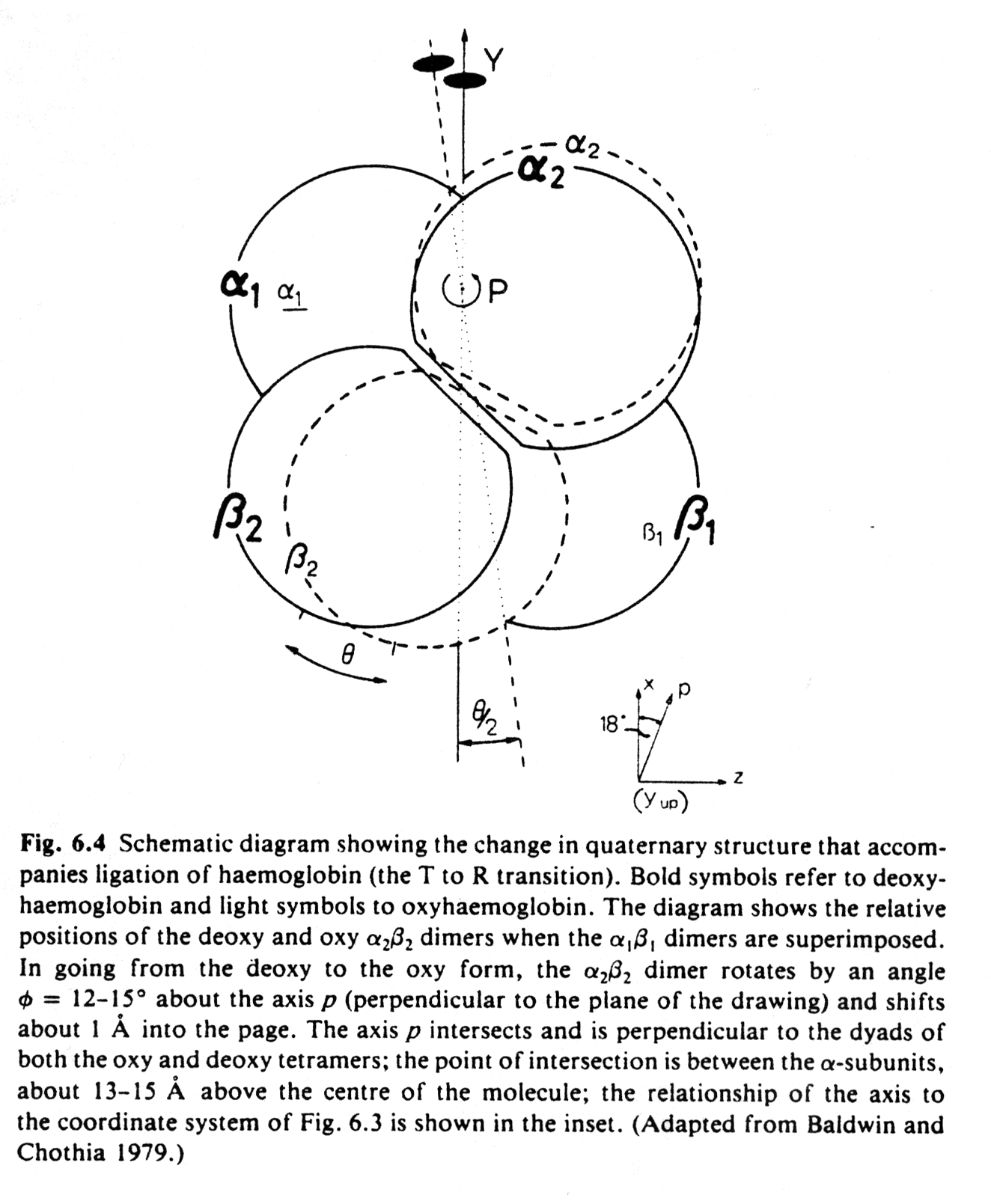
It is the movement of the Fe(II) towards the heme plane that triggers the T to R conformational change. The relative displacement between subunits has only two stable conformations, i.e., the T and the R state. Intermediate positions are energetically unfavorable, therefore the change in quaternary structure provides two conformational endstates, one with a low O2 affinity, the other with an increased affinity to the heme groups. These two states are stabilized by two energetically equivalent sets of hydrogen bonds at the subunit interfaces. After the binding of the first oxygen, all subunits are converted from the t to the r state whether or not they bind a ligand.
Fig. The hydrogen bond contacts at the a1b 2 interface (Fermi, p.180)
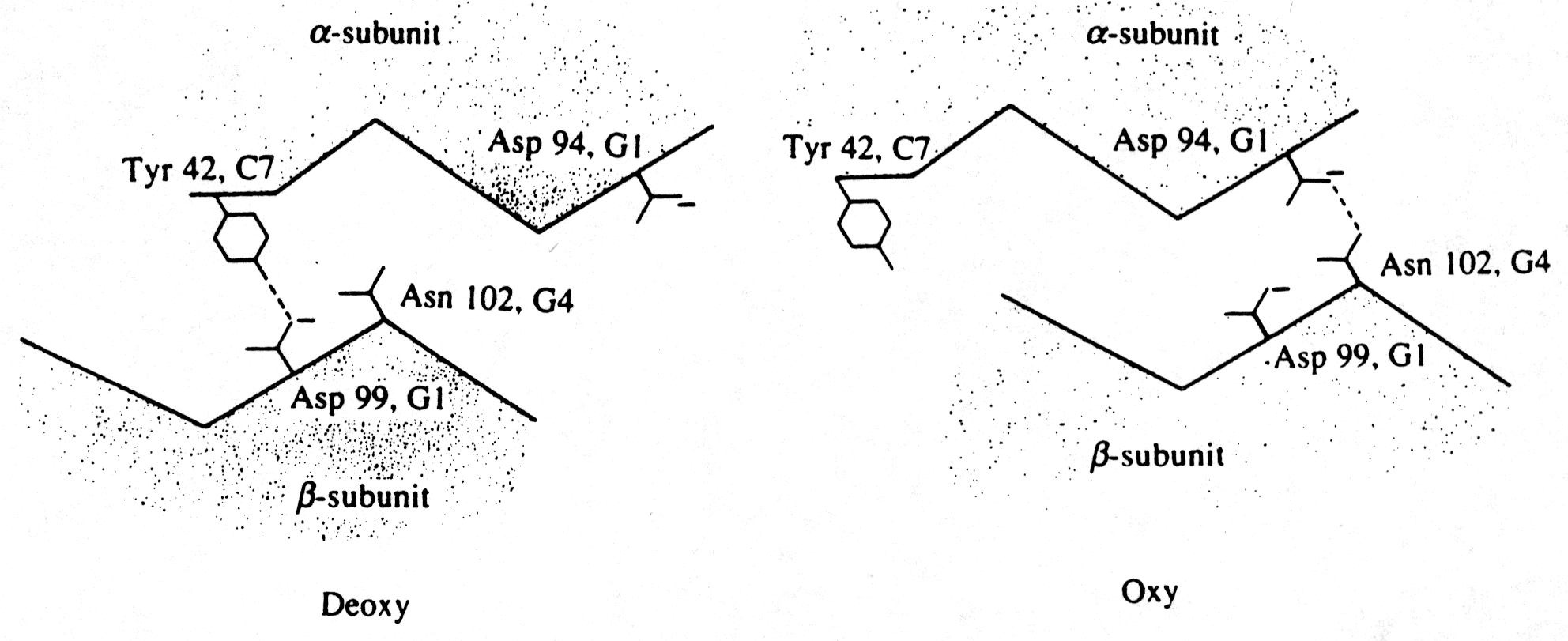
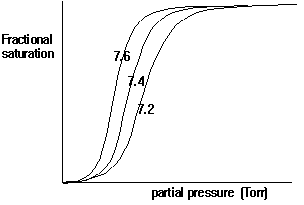
In general, cooperative interactions occur when ligands bind to proteins or protein complexes. If the binding of ligands is regulated by different ligands at sites other than the heme, then we call this allosteric interaction. Ligands function as effectors or modulators at different sites in the protein. If the ligands are identical, this is known as homotropic effect, if they are different, it is known as heterotropic effect. Molecular oxygen is a positive, homotropic effector for hemoglobin. The protein also exhibits heterotropic effects which induce mostly negative cooperativity, i.e., they decrease the affinity of O2 to hemoglobin. For example the generation of CO2 in the muscle and its conversion into bicarbonate releases protons. These protons bind to Hb and reduce its affinity for O2 by stabilizing the T state. This effect is known as Bohr Effect. In addition, CO2 binds reversibly to hemoglobin at its NH2 terminal end to form carbamates (R-NH-COO-). CO2 binds better to the N-terminal end of deoxyHb, and once bound, stabilize the T state.
Fig. pH dependence of oxygen binding to hemoglobin
Oxygenation changes the electronic state of the Fe-heme complex, a change
that can be measured spectroscopically as a slight shift from purple to
red. O2 binding is competed by CO, NO, and H2S.
In hemoglobin this binding is stronger than that of O2 causing
death by quickly binding to the deoxygenated hemoglobins.
5. The symmetric model of allostery in protein complexes
The symmetry model of allosterism describes the cooperative binding of ligands to an enzyme complex. It was developed by Jaques Monod, Jeffery Wieman and Jean-Pierre Changeux. It is therefore also called the MWC model of allosterism and is defined by the following rules:
- Each subunit exists in two (or more) conformations t and r, they are in equilibrium whether ligands are bound or not.
- The ligand can bind to a subunit in either conformation, the conformational change alters ligand affinity.
- The molecular symmetry is conserved during conformational change.
Subunits therefore change in a concerted manner, there are no oligomers
containing both t and r states.