Protein Structure: a -helix, b -sheet and turns
1. Hierarchical Structure of Proteins
Amino acids are covalently linked through peptide bonds to form linear
polymers called peptides or proteins. These linear polymers can spontaneously
fold into three dimensional structures, called native fold, i.e.,
a biologically active form. The structure of proteins (and peptides) can
be analyzed on 4 different levels. These four levels are summarized in
the table below:
| I | primary | linear sequence | 1°-D |
| II | secondary | local, repetitive spatial arrangements | 2°-D |
| (II/III supersecondary; motif) | |||
| III | tertiary | three dimensional structure of native fold | 3°-D |
| IV | quaternary | non-covalent oligomerization of subunits (single polypeptides) into protein complexes | 4°-D |
2. Peptide bond formation
Two amino acids can undergo a condensation reaction, where the carboxyl group reacts with the amine group. The formation of this peptide bond (see box in figure) produces a dipeptide and a H2O molecule, exhibiting a dipole moment m = 3.7 Debye for the peptide bond (arrow in figure). Note that the dipole moment of the dipeptide is different from the peptide bond dipole moment because of its charged amine and carboxyl groups.
Fig. Chemical structure of a dipeptide
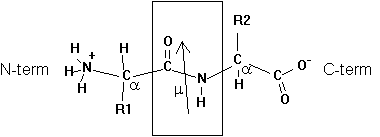
Adding additional amino acids to the growing peptide chain (always form N-term to C-term direction in living cells) produces a polypeptide chain or protein. The order in which amino acids are linked to the growing chain defines its primary structure or sequence.
The geometrical character of the peptide bond is that of a rigid plane between the two flanking a -carbon atoms. The reason for this structural stability (there is no rotation around the C-N bond) can be explained by the electronic resonance character of the O=C-N structure. The double bond character changes between the O-C and C-N bonds.
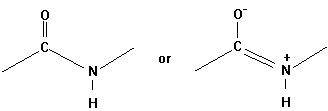
Evidence for this structure comes from X-ray crystallographic studies
of simple peptides showing that the N-Ca bond
length is 1.46Å as expected for a single bond. The C-N peptide bond
is 1.33Å long, only a little longer than the value of 1.27Å
for the average C=N bond length in model compounds. Similar X-ray studies
show the six atoms Ca NHCOCa
very close to being co-planar (Pethig, p.41).
3. Secondary structure
Immersed in water a polypeptide chain will not stay in an elongated form, but fold up according to the polarity of the side chains it contains and the rotation of peptide backbone bond angels largely determined by Van der Waals radii of side chains.
To understand protein structures we can measure two torsion angles ( also called dihedral angles) in the backbone which define the tilt between two neighboring amide planes (the plane of the peptide bond) with the Ca at the center of rotation:
F Ca¾ N Y Ca¾ C
Thus we can calculate the allowed backbone conformations of a peptide
through interplay of rotation around the bonds defined by the torsion
angles Phi (F ) and Psi (Y
) and the steric hindrance of side groups determined by their Van der
Waals radii. The resulting conformational map is called Ramachandran
plot (after Ramachandran, who invented it). A free rotation (no energy
needed, due to thermal motion) around a C-C bond is possible in the absence
of any steric (i.e., Van der Waals radii) constraints. (Note that the
peptide bond torsion angle w has a fixed value
and can be neglected in a Ramachandran plot.)
Fig. Peptide backbone and torsion angles
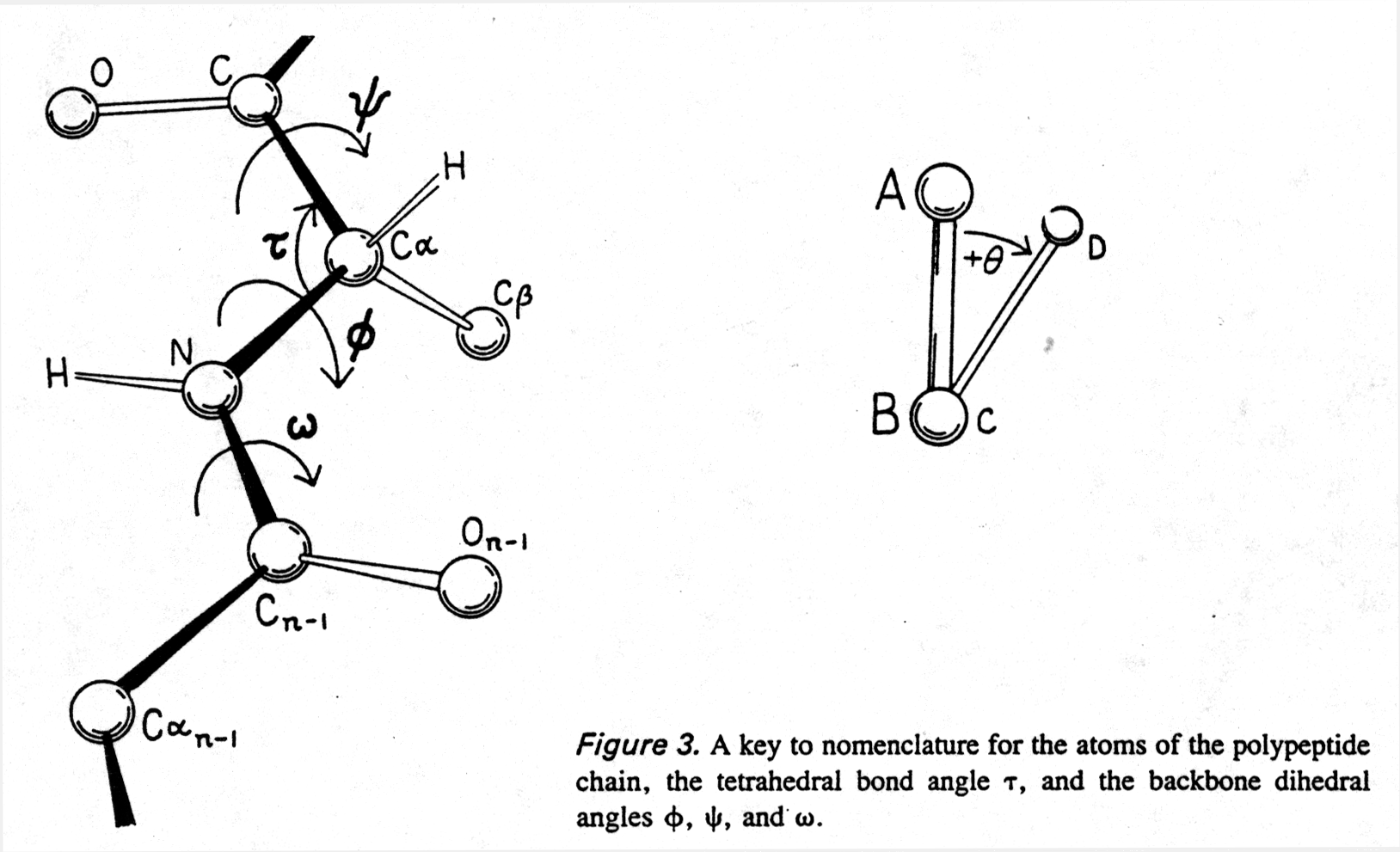
from Richardson and Richardson, pp.4/5
Fig. Ramachandran plot for all residues except Gly and Pro
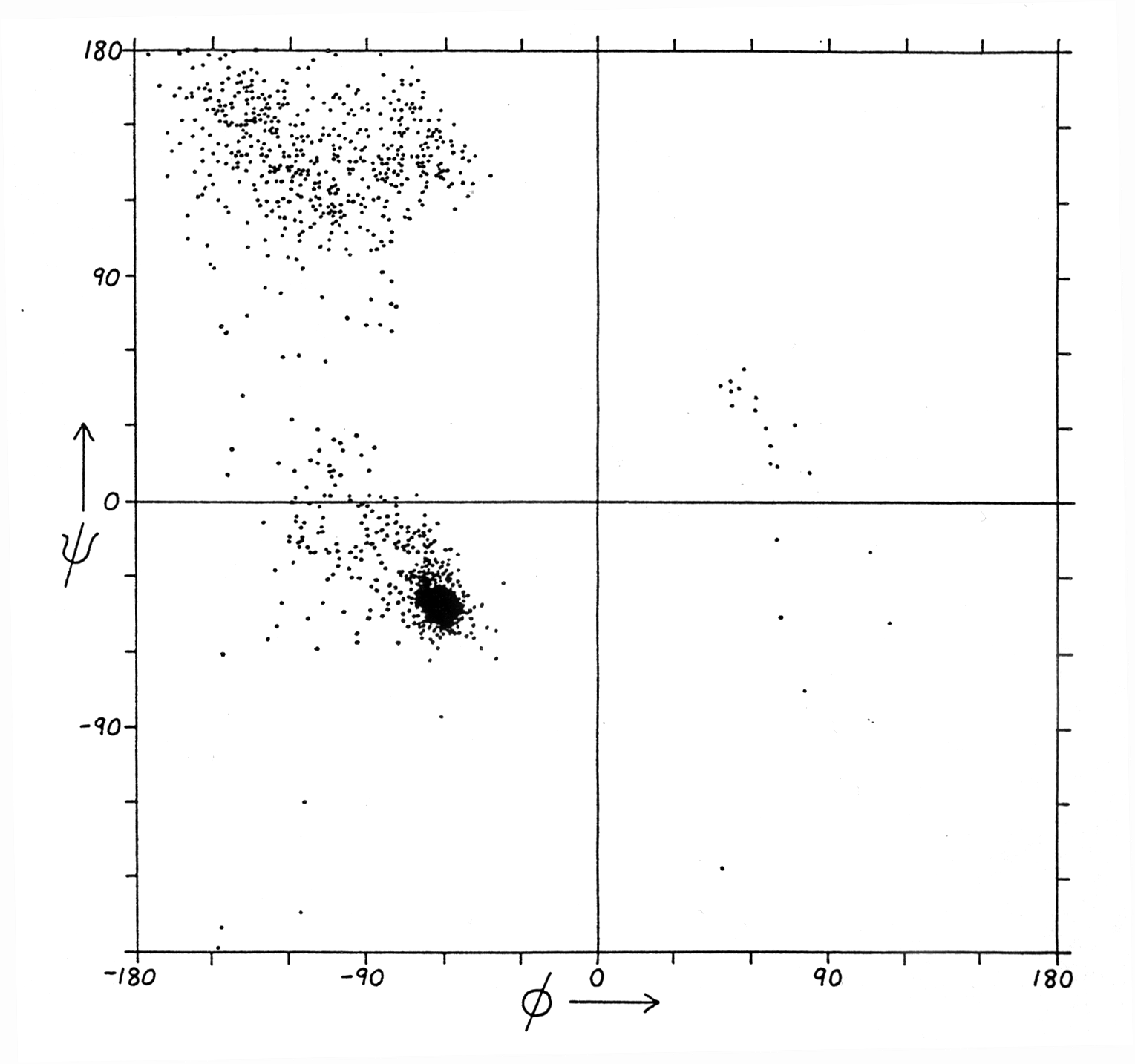
Note: values come from structure data sets
(from Richardson and Richardson, p. 6)
Ramachandran plots for proteins show three confined regions of the conformational map which are physically accessible to them. The Ramachandran plot of a protein is a full description of the polypeptide backbone conformation (excludes side chain conformations). Three important regions of the Ramachandran plot describe the most commonly found secondary structures in proteins:
a -helix : a right handed helical structure with average torsion angles F =-57 and Y =-47
b -sheet: parallel (F =-119 and Y =113) or anti-parallel pleated sheet structures
b -turn: minimal loop structures of 3 to 4
amino acids with defined torsion angles
4. The a -helix
The a -helix is one of two secondary structures (the other being the b -sheet) predicted and discovered by Linus Pauling in 1951. It is a right-handed helix with the following spatial parameters:
F = -57°
Y = -47°
n = 3.6 (number of residues per turn)
pitch 0.54nm (or 5.4Å)
The helix has a specific hydrogen bonding pattern, where the backbone C=O group of residue n bonds with the N-H of residue n+4. The atomic distance between the N and O measures 0.28nm. The H-bonds are almost parallel to the helix axis and the total dipole moment gives the helix a dipole moment that points from the N-term (+) to the C-term (-). This helix dipole is important in the interaction of neighboring helices in the packing of secondary structural motifs into the 3-D structure. An example of the functional importance of a helical dipole moment is shown in section 2.3.
Fig. Orientation of dipole moment(s) in an a -helix as defined by chemists (m +® -)
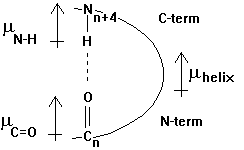
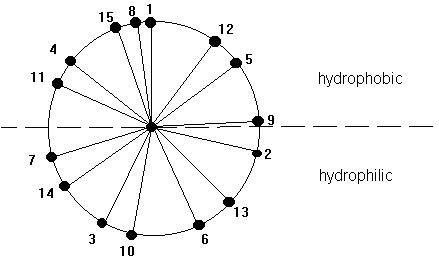
The core of the helix is packed. The backbone atoms are in Van der Waals contact with each other across the helix axis. A helix can be represented in its so called wheel presentation. A helical wheel is a projection in 2-D along the helix axis and displays the orientation of the side chains on a 360 degree map with respect to the side of the helix. This wheel presentation is helpful for the detection of potential amphipathic helices. Amphipathic helices have a polar and a non-polar side and this plays a crucial role in helix-helix interaction and in the interaction of small peptides that have a helical conformation with the interaction with membranes, air-water interfaces, and self-assembly processes.
Fig. Helical wheel representation of a 15 amino acid long a -helix
5. The b -strand and b -pleated sheet
In 1952, Pauling and Corey predicted the b -pleated sheet structure as an alternative secondary structure to the a -helix in proteins. b -strands are elongated peptide segments with atomic distances from side chain n to side chain n+2 of 0.7nm.
Fig. b -strand
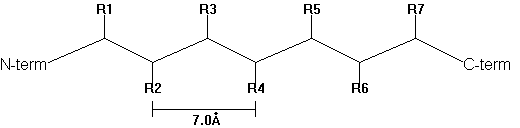
Single b -strands are not stable structures but occur in association with neighboring strands. Thus they can be found as either parallel or anti-parallel with respect to the N- to C-terminal direction of the adjacent peptide strands.
anti-parallel N® C
parallel N® C
C¬ N
N® C
Like a -helices, b
-pleated sheet backbones are fully hydrogen bonded, but here the H-bonds
occur between neighboring strands (intermolecular). The H-bond geometry
is different in the parallel and anti-parallel conformations (see also
section 1.8).
Fig. Hydrogen bonding in antiparallel b sheet
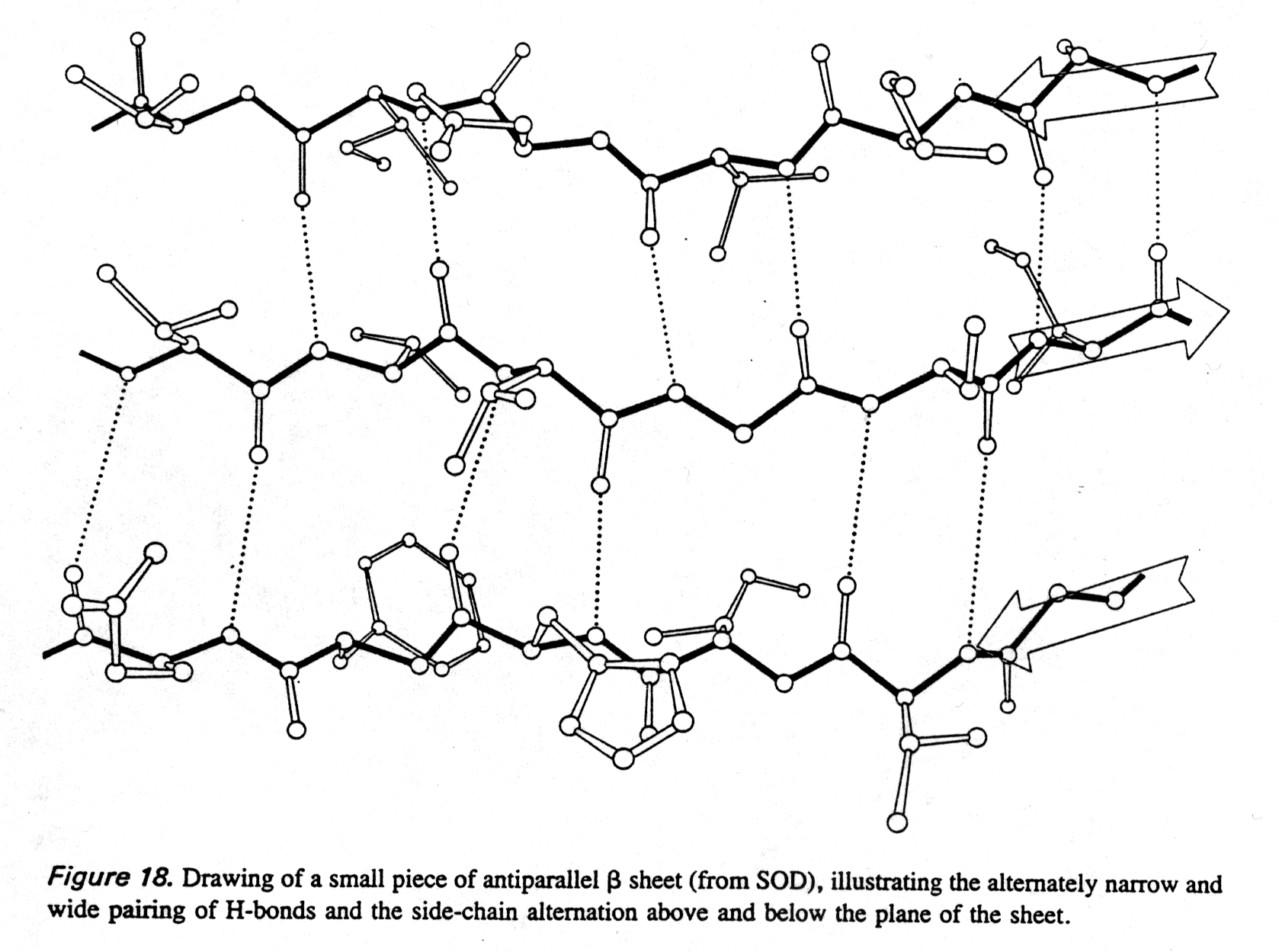
from Richardson and Richardson, p.18
More than two strands can form into sheets which form extended right-handed twists. Such extended b -pleated sheets (called super secondary structures) can often been found in the cores of proteins. Alternatively, bundles of 4 closely packed a -helices (so called a -helical bundles) are also found at the center of globular proteins.
If the b -strand contains alternating polar
and non-polar residues it forms an amphipathic b
-sheet. This distribution of hydrophilic and hydrophobic residues
has been observed in the membrane protein porin that forms a b
-barrel structure (section 2.3), where the non-polar residues stick into
the hydrophobic part of the lipid membrane and the hydrophilic residues
form part of the channel interior responsible for the passage of small
molecules across the membrane.
6. The b -turns
To combine helices and sheets in their various combinations, protein structures must contain turns that allow the peptide backbone to fold back. Two turn structures will be discussed here using their Ramachandran plot coordinates:
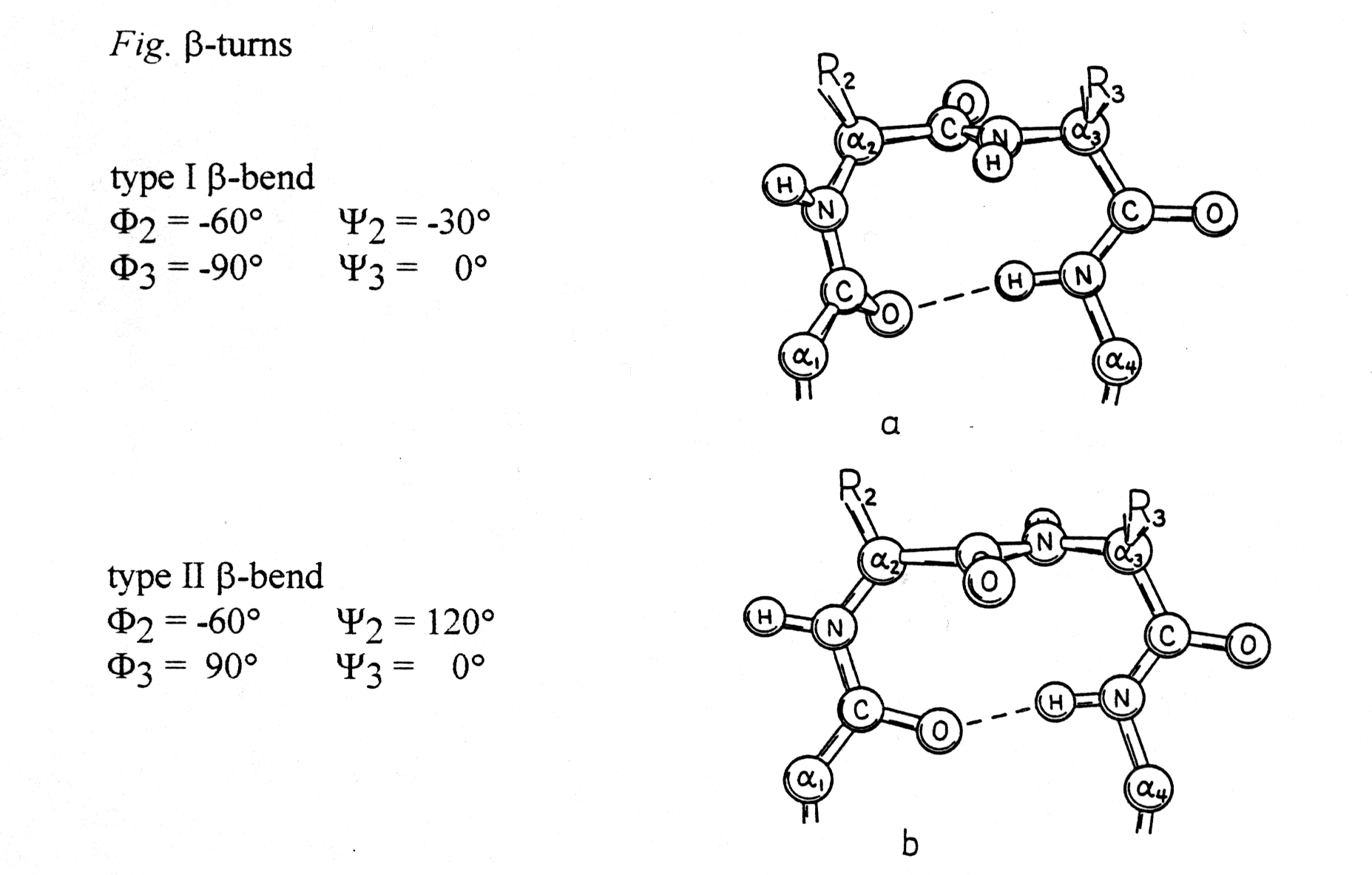
from Richardson and Richardson, p.24
These turns can be found almost always on the surface of proteins and
often contain Pro and/or Gly. Proline gives the backbone a special rigidity
(fixed Phi torsion angle at -60° , Ca
-N) and glycine has a high flexibility because of its hydrogen substituent.
Turn structures are also stabilized through H-bond formation.
7. Super Secondary Structures or folds
There are many structural motifs that are formed by the combination of
a -helices and b
-strands. They can be classified in groups as shown below (see also section
3.9). The 4 most common (among >300) motifs are:
1. bab2. b -hairpin (or anti-parallel b -pleated sheet)
3. aa (the most common member being the 4 helical bundle)
4. b -barrels (see Greek key motif)
All these motifs can be found in many different proteins and enzymes that
have very diverse functional and structural properties. a
and b -structures are often found in combination
as seen for carboxypeptidase and trios phosphateisomerase
and for pyruvate kinase as shown in the figure below.
Fig. The three domains of pyruvate kinase (PDB entry 1A3W)
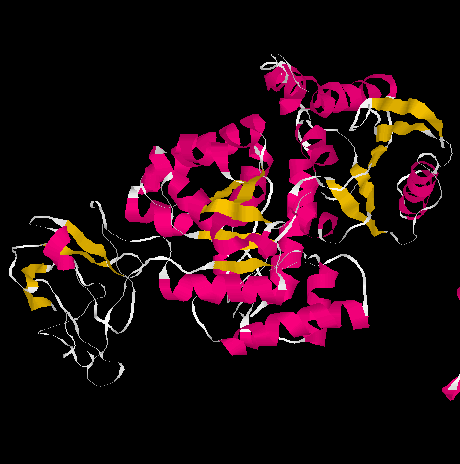
The three domains are shown separately below:

from Richardson and Richardson, p.29
This motif classification can be important in finding protein domains
and study protein structure evolution. The functional variability
associated with homologous motifs, however, makes the classification of
proteins according to their functional properties more useful.
8. Tertiary structure
The tertiary structure is the 3 dimensional, native structure of a single polypeptide or protein. A protein normally is folded into a compact structure, usually referred to as 'globular protein', a term traditionally associated with water soluble proteins. The secondary structures are stabilized by the final, native fold. The nativefold is defined as the active conformation.
Amino acids are the components of proteins and their sequence determines the native fold. Of course this is always in combination with the solvent, in this case water. The location of an amino acid in a protein fold correlates with the energy of hydration of each individual amino acid residue and the entropy of the side chain, polypeptide backbone, and solvent molecules (see section 2.7). As mentioned earlier, polar and charged amino acids are likely to be hydrated, whereas the non-polar residues stick to each other and often form the core of a protein, forming a usually hydrophobic core that stabilizes the fold of water soluble, globular proteins. For membrane proteins exhibiting both hydrophobic and hydrophilic surfaces, the amino acid distribution is different from globular proteins but the same rules of the hydrophobic effect apply. Here is a list of the distribution of amino acids in globular and membraneproteins:
Table Distribution of amino acids in proteins
| Residue | Globular protein | Membrane proteins |
| non-polar V, L, I, M, F, Y, W |
in interior, hydrophobic core | surface, lipid anchor |
| polar, charged R, K, D, E, H |
surface, in interior function often as catalytic sites | extra membranous, electrostatic interaction, functional group of channel proteins, forming a hydrophilic core |
| polar, neutral S, T, N, Q, Y, W |
surface & interior (H-bond network) | surface, inside part of channel, H-bond network |
The following list is a selection describing the special roles of amino acids in protein structures (from Richardson&Richardson, pp.43-75): .
Proline: The special consideration of proline is its role in a -helices of membrane proteins because proline induces a 30° kink that has been related to functional aspects in transport processes. Proline is frequently found in turns, non-repetitive structures, ends of a -helices, and is almost always exposed at the surface of globular proteins, where, due to the non-polar characteristic of its ring structure, it exhibits a hydrophobic spot. Proline does not fit into the regular part of either helix or sheet structures because it does not have a backbone-NH available to take part in an H-bonding and not because of its restricted torsion angle values (F =-60° ; Y =-55° or 145° ). In the helix center, the ring pushes away the preceding (N-terminal) turn of the helix by ~1Å producing a 30° bend and breaking the next H-bond as well.
Cystein: It can form disulfide bridges, the only known polypeptide linkage with a non-linear topology (for an exception see peptidoglycan, section 1.7) when the thiol groups are oxidized. It also binds Fe in Fe-S clusters and other prosthetic groups (necessary for enzyme activity) including Zn, Cu, and hemes. Cystein is poor at H-bonding.
Tyrosine: Depending on its position in a protein, the hydrophilic or hydrophobic part of the aromatic ring structure prevails. In general, polar groups like -OH do not significantly affect hydrophobic effects experienced by the main part of hydrocarbon chains (this is of course not true for small alcohols). The phenolic group of this amino acid, together with Phe and Trp accounts for most of the UV absorbance and fluorescence behavior of proteins. Spectroscopists use it as a tool to study the folding state of a protein or simply monitor its abundance.
Fig. Proline in center of a -helix;
stereodiagram
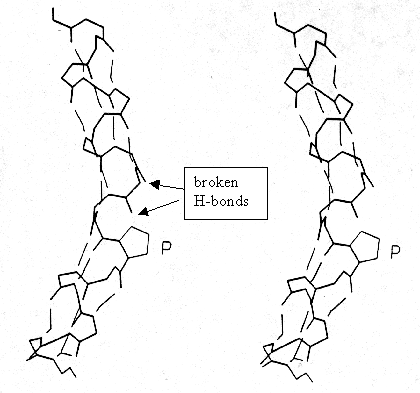
from Richardson and Richardson, p.51
Histidine: Having a pKa = 6.0, the imidazolium group barely ionizes at physiological pH. It is therefore only partially charged (+ 8% at pH=7) and can participate in enzymatic reaction that involve the exchange of protons. It is worth mentioning that pK values are not absolute, but change with the dielectric property of the solvent (dielectric constant of the protein surface or protein interior, charged groups located nearby etc.). This is a major challenge for understanding structure-function relationship.
Serine and Threonine: These two amino acids are involved in H-bonding. In a -helices, Ser can H-bond to the C=O of residue n-3 and is thus a helix breaker because it shares this H-bond with the intramolecular N-H .. O=C of the helix backbone. In the first turn the serine acts as a cap structure (see Fig. of BPTI on in section 2.1.8 for details). Ser has a high frequency of occurrence in turns, non-repetitive structure and H-bonds to neighboring backbone N-H and C=O. Thr is commonly found in amphipathic anti-parallel b -sheets. The -OHs are reactive and can be modified by chemical linkage to phosphates (phosphorylation) and oligosaccharides (glycosylation), both common post-translational modifications of proteins (section 3.5).
Aspartate and Glutamate: Acidic amino acids, they bind Ca++ providing six coordination sites, as shown in E-F hand structures (calmodulin, section 2.5). They are commonly found on the protein surface and involved in protein-protein interaction. Buried in the hydrophobic core, they stabilize the protein structure if ion pairs are formed, mostly with Arg.
Lysine and Arginine: Mostly exposed at surface, solubilize
proteins and interact with nucleic acids' negatively charged phosphate
backbone. The long side chain of lysine is highly flexible and due
to its e -amine group a great solubilizer
of globular proteins. Arg is less flexible and provides 5 H-bond donors
in its large, rigid planar array of the guanidinium group. Optimally
forms ion pairs with Asp and Glu, or peptide C-terminal ends.
m2 = 2DoMkTd /NA
where d is the dielectric weight increment, M the molecular weight of the protein, Do the dielectric constant of water. The dielectric weight increment d can be calculated from the dielectric constant of the protein solution (D), water (Do), and the protein concentration (c):
d = (D - Do)/c
The following table lists the protein dipole moments for a few proteins
as given by R.Pethig, p.86 (Table 3.4)
| protein |
m (Debye) |
| horse carboxy hemoglobin |
|
| myoglobin |
|
| horse myoglobin |
|
| egg albumin |
|
| horse serum albumin |
|
the listed proteins are rich in a -helices.
Single polypeptides can associate with each other to form larger protein complexes of geometrically specific arrangements, called quaternary structures. Individual polypeptides in protein complexes are referred to as subunits (and should not be confused with domains). We will encounter many examples of quaternary structures or subunit composition of protein complexes. Most enzymes are complexes of proteins and the symmetry and stoichiometry of the composition of the complexes is crucial for their activity.
We can distinguish two different compositions, the homomeric and
heteromeric complexes. Heteromeric composition of most protein
complexes gives the cells an additional level of variability and complexity
it can use for its activity. Often, heteromeric compositions of protein
complexes are tissue specific or developmental specific and multiple
genes can control the activity of a single heteromeric protein complex.