|
Table of contents II Determining physical properties of membrane
proteins (updated 8/43/06)
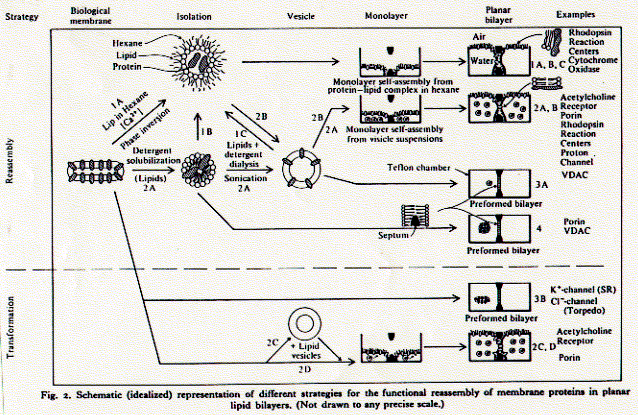
4. Analyzing protein structures and proteomics techniques There is a simple experimental need to isolate and purify [membrane] proteins. Biological systems are complex and it is normally difficult to impossible to determine the structure function relationship of a protein in its native environment (in vivo). As a first step, therefore, proteins are reconstituted into simpler systems (in vitro) by removing them from their native surroundings. This allows the determination of intrinsic properties of a protein, properties which are largely independent of other membrane components; molecular weight, charge, secondary structure composition, and often also its function as receptor, pump, transporter, or channel. Properties of proteins studied in reconstitution systems are called intrinsic because they depend on a minimal set of other molecules; a protein in aqueous solution, or if it is a membrane protein, a detergent extract. The ionic strength, pH, temperature and protein concentration comprise the few variables. An in vitro system thus has clearly defined parameters and the measurement of macroscopic properties such as heat capacity, UV absorption, circular dichroism spectra can easily be interpreted for moclecular structure and mechanisms. Once certain properties have been established as being 'intrinisic' to the protein, its function can be studied in vivo and the influence of other cellular components on its activity determined. Fig. Outline of reconstitution pathway for ion channels Critical micellar concentration While it is impossible to keep membrane proteins in a polar solvent,
their are many ways to isolate them from their native membrane environment,
purify them to homogeneity (remove all other proteins), and reconstitute
purified proteins into supra molecular structures mimicking either solubility,
or orientation, or both properties of membranes. Removing or isolating
membrane components from native membranes is done with the help of detergents.
Detergents are small amphipathic molecules that have a propensity to
form micelles in aqueous solution. Micelles are small, spherical particles
of a few dozens to hundreds of monomers with a hydrophobic core and
a hydrophilic surface. Detergents are found in three phases all of which
are in chemical equilibrium: - free monomers
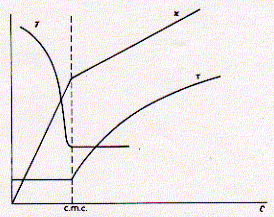
This figure shows typical behvior of three solution properties as a function of detergent concentration. As can be seen, the CMC delineates discontinuous behavior in isotherms for the surface tension g, specific conductivity k, and turbidity t. For all three properties, the concentration dependence changes when the detergent concentration surpasses the critcal micellar concentration. The surface tension decreases with increasing concentration of the surfactant indicating surface excess and monolayer formation. Above the CMC, however, the surface tension no longer changes, because the free monomeric concentration of the detergents remains constant. At any given concentration, detergent molecules are in euqilibrium between monolayer, monomeric solute, and micellar component. Only the number of micelles increases with increasing concentration of detergent above the criticall micellar concentration. This is evident by the increase in turbidity (light scattering) of the solution. If the dergent carries a [positive or negative] charge, the conductivity of solution increases with increasing free detergent molecules. The observed discontiunity at the critical micellar concentration is itself a function of the size of the micelles formed. It is imporant to keep in mind that micelles have a defined size and aggregate number (number of monomers in micelle) and don't increase with increasing concentration, if the concentration does not dramatically exceed the CMC. The size and aggregate number of micelles, however, can be well characterized for different chemical structures of detergents. The concentration above which micelles start to form is a function of monomer structure and chemical composition. The CMC decreases with increasing chain length of the apolar residue. The CMC decreases by about a factor of 0.3 when the hydrocarbon chain length is increased by two carbon atoms. Micelle size also increase with an overall increase in ionic strength of the solution. The observed conductance of ionic surfactants above the CMC is lower than for the monomeric species. This is mainly due to the presence of counter ions which interact more strongly with the more structured multiple charged micelle surface effectively shielding this charge. Micelles are only about 20% charged as compared to the monomeric species. This must clearly be an effect of supra molecular structure formation and charge-charge interaction between head groups as well as counter ions. Table List of some commonly used detergents
for membrane protein studies
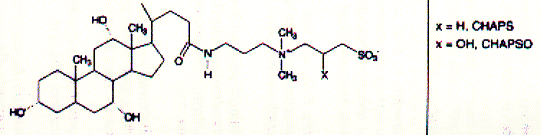
Fig. Structures of CHAPS (top) and Triton detergents Chaps and Triton X-100 are two very commonly used mild detergents for membrane protein purification. Unlike SDS ( a sulfonated alkyl unit) they do not denature most proteins and even leave quaternary or larger aggregate forms intact. Light scattering The size of ionic micelles as characterized by the number of surfactant molecules per micelle can be measured by light scattering experiments (van Holde, chapter 7.1), which allows a determination of molecular weight of particles in solution without specific information about their structure. Since visible light is electromagnetic radiation, the electric component of the wave should produce oscillation of the electron distribution of the targeted sample molecule. Electron oscillation induces dipoles which absorbe light energy while dispersing this energy in directions other then the direction of the incoming radiation. The oscillating dipole moment is a function of the molecular polarizability a of the molecule with the dipole moment m being the product of the polarizability a and the field strength E of the radiation. m = aE In general, we are inerested in the change in intensity that occurs in the direction of the incoming radiation I/i. This loss in intensity is proportional to the degree of scattering which is a function of the number and size of the particles (here micelles) in solution. Light scattering from a number of small particles (3-10 nm) as compared to the wavelength of the radiation (400-800 nm) is known as Rayleigh scattering. For all practical purposes, the polarizability of a molecule is a microscopic property and not easily determined for biological macromolecules. Rayleigh scattering theories related change in radiation intensity with the refractive index of a solution, a macroscopic property which is proportional to the polarizability as shown below. A convenient measure of polarizability for visible light scattering is the square of the refractive index n: n2 - n02 = 4pNa n is the refractive index of the solution, n0 the refractiv index of the solvent, and N the number of particles with polarizability a. It can be shown that scattering produced by a solution containing a weight concentration C of particle of molecular weight M depends on the product CM and light scattering measurements (Rayleigh scattering) not only indicate trubidity but can be used for molecular weight determination.
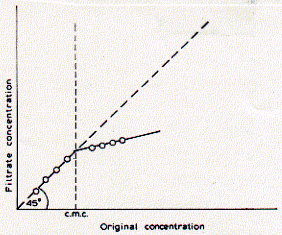
Distribution of micelles sizes is relatively homogeneous for any given detergent and the number of average aggregation numbers exhibits a narrow distribution. The current model of the structure of ionic micelles of synthetic surfactants in dilute aqueous solution is that of a rough-surfaced sphere or ellipsoid containing 40-150 monomers. The detergent most commonly used for the isolation of membrane proteins for analytical purposes is sodium dodecyl sulfate (SDS). It is a negatively charged, sulfar containing surfactant with a C12 carbon residue with a critical micellar concentration of 8.1mM. Phospholipids are weak surfactants and have a very low CMC value because of their two long fatty acid chains. Due to their structure of a small head group with a large, bulky hydrocarbon part, phospholipids would ideally form elongated elliposidal structures. In fact, the ideal aggregate structure of phospholipids above their CMC are extended two dimensional sheet structures -- phospholipid bilayers. Phospholipids, however, solubilize very well in dergent micelles made from SDS. The CMC for phospholipids has been measured by filtration techniques,
assuming that the phospholipid conentration of a solution pressed through
a paper filter absorbes more lipids in the aggregated form than the
free monomers in solution. Indeed, a discontinuity can be observed when
comparing the phospholipid concentration of the start solution with
that of the filtered solution above a certain concentration.
Below the CMC, original conentration and filtrate concentration are
equal, while above the CMC, much of the phospholipids are retained in
the filter because the 'micelle' aggregates get stuck in the pourous
material. The CMC for DPPC (dipalmitoylphsophatidylcholine) has been
determined in pure water as 0.5nM. The CMC increased when increasing
the molar ratio of methanol of the solution and reached 10mM in pure
methanol indicating good solubility of phospholipids in this organic
solvent. Naturally occuring phospholipids are thought to have CMC values
in the range of 0.01 to 10nM. This is six orders of magnitud lower than
the CMC values for common detergents used for membrane [protein] solubilization
(for a further discussion on phospholipids see section on vesicle preparation).
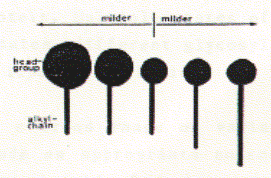
Isolation and purification of membrane proteins There are many different detergents available for the solubilization of membrane proteins. Different classes of detergents are useful for different purposes and different membrane proteins. The most important factor in choosing a detergent is its effect on the protein structure. SDS is a strong anionic detergent which is primarily used for analytical procedures because it not only solubilizes membranes by forcing proteins and lipids into its micellar aggregates, but it usually denatures proteins by interfering with the hydrophobic packing of the core amino acid residues. In fact, SDS is the detergent of choice for analytical polyacrylamide gel electrophoresis (SDS-PAGE). For a protein isolated for functional studies, milder detergents have to be selected. A mild detergent is detergent that by definition does not cause a protein to denature or cause loss of function in vitro. The classes of detergents that compirse mild detergents are often non-charged and tend to have small aggregation numbers. In addition, detergent with small headgroup and long alkyl chains, and those with large head groups and short alkyl chains tend to be milder as shown here. See also table above. Fig. Correlation between head group
and alkyl Some membrane proteins are so sensitive to detergent interaction that any loss of phospholipid-protein interaction causes a loss of function. The nicotinc acetylcholine receptor is one such protein. Other proteins are resistant against detergent denaturation, even in the presence of SDS. The outer membrane pore forming protein porin is a stable protein. It is a homotrimeric protein which only dissociates into its individual monomers in SDS at temperatures above 70 degrees Celsius. The SDS polyacrylamide gel below shows the result of a purification procedure of the general diffusion porin OmpF from Escherichia coli. The gel includes from left to right different fractions and samples of extracted E.coli membranes with increasing accumulation and puritiy of the OmpF porin. The native membranes (lane 1) contain a complex mixture of proteins, visualized by the many bands separated over the entire molecular weight range of the gel. With increasing purity and removal of undesired proteins by gel filtration and ion exchange chromatography, only a single protein species is left. Fig. Purification of E.coli OmpF porin Coomassie blue staining of proteins in polyacrylamide gels reveals
the banding pattern of proteins. This is due to differences in molecular
weight, quaternary structure (complex formation), or post translational
modification.
Principles of gel electrophoresis The principle behing the separation of proteins of different molecular
weight by polyacrylamid gel electrophoresis (PAGE) is based on the movement
of charged particles in the force field of an electric field. This can
in generally be treated as a transport process. Molecules can be separated
on the basis of their - mass
fo = 6phR with fo being the frictional coefficient and R the [Stoke's] radius of the particle. There exists no good theory for polyacrylamide gel electrophoresis, because the moving particle is immersed in a polymer matrix soaked in an electrolyte with SDS, in other words, a non-ideal solution. For the movement of a charged particle in an electric field, Coulomb's law is a good approximation for the force working on the protein: F = zeE with z the number of charges, e the unit negative charge of a single electron, and E the electric field strength in the gel. Since the electric and frictional force are equal at equilibrium (constant velocity of moving particle), the following relationship holds: fn = zeE The electrophoretic mobility U is defined as n/E = ze/f. For spherical particles the frictional coefficient can be replaced with Stoke's law:
z e Proteins are not separated due to their native charges alone, but strongly
associate with SDS molecules in an equal weight to weight ratio. Proteins
in SDS electrophoresis gels therefore carry negative charges proportional
to their size or molecular weight. The mobility is linear proportional
with the logarithm of the molecular weight. See the molecular weight
standards in the above polyacrylamid gels. 2D Gel electrophoresis The charges of the amino acid residues are of course not umimportant and the net charge of proteins depends on the pH of solution. This pH dependency is used to determine the isolectric point of proteins and for protein separation techniques is known as isolectric focussing. Here, the proteins are separated in an electric fiel along a pH gradient. The proteins change their net charge and exhibit different charge/weight ratios as they move along the pH gradient. Thus a second dimension can be applied to separate proteins; the first dimension is the SDS associated charge/molecular weight separation; the second dimension the isolectric focussing of proteins of equal size in a pH gradient. Fig. Schematic diagram of protein separation on 2D gel electrophoresis
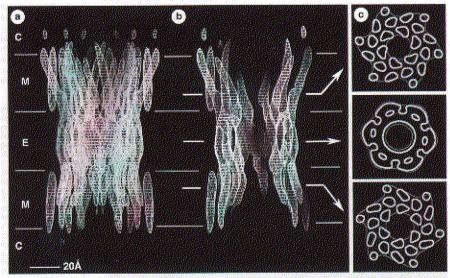
2D gel electrophoresis allows the separation of proteins of equal molecule weight but different charges in a pH gradient. Although developed in the early 1970s, 2D gel electrophoresis was not widely used because of the complexity of the banding (or spot) pattern on these gels and the limited ability to unabmiguously identify which proteins belongs to which spot on a gel. Technical information on isoelectric focussing (pH gradient dimension) and molecular weight dimension can be found at Expasy. Studying protein complex assembly - Sedimentation Porins are found in large copy numbers in the outer membrane of Gram-negative bacteria. A single E.coli cell contains as many as 105 units. From E.coli strains containing more than one porin gene like ompF, phoE, lamB which code for the general diffusion porin, the anion specific phosphoporin, or the maltose specific maltoporin, it can be shown that the porin content of the outer membrane can be changed in 10 to 20 minutes, demonstrating that these seemingly stable structures are highly dynamic. This is even more remarkable considering the fact that porin trimers are found in close proximity in the outer membranes, essentially forming a densly packes two dimensional moleculer sieve. This tendency of porin to form large aggregates or ensembles in the plane of the membrane, made its isolation and purification relatively straight forward, and also allowed the determination of its structure by electrons microscopy. Removal of phospholipids (and lipopolysaccharide) from outer membrane fragemnts results in almost pure, two dimensional porin crystalls. Bacterial membrane proteins are not unique in forming this native two
dimensional sheets. Also muscular nicotinic acetylcholine receptor and
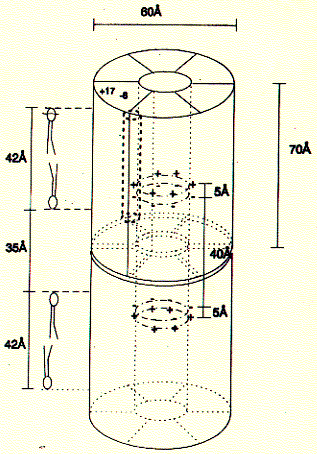
gap junction channels are found in Although no high resolution structure has been solved yet, a 7.5 angstron resolution from electron diffraction studies has been obtained and largely confirms the topology diagram discussed above. Fig. 7.5 Angstrom map of a heart muscle gap junction (connexin
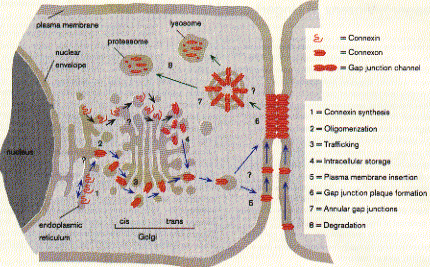
43 complex) The structure from electron diffraction studies of recombinant connexin 43 complexes shows the electron densitiy profile of an entire channel outlining the mostly alpha helical organization of the transmembrane spanning domains (M) and the molecular interface across the inter cell gap (E). The full view has been reduced to show the channle interior (panel b) and cross section of the image reconstruction map are shown in panel C. This large protein complex spanning two cell membranes is particularly interesting in studying not only the structure function relation ship of the native channels, but also their synthesis, membrane assembly of a connexon in each cell and connexon-connexon assembly across the narrow gap of about 2nm between coupled cells as well as the large 2D sheet structures (plaques) that are formed in specialized membrane regions. . Fig. Proposed assembly pathway for gap junctions The figure above outlines the synthesis, assembly and intracellular transport pathway for gap junction channels in a hypotheticla mammaliona cell. Eight different stages of the process can be separated and studied independently -- synthesis, oligomerization, trafficking, intracellular storage, plasma membrane insertion, plaque formation, and two steps of degradation -- annular gap junctions and their [proteolytic] degradation in lysosomes and proteasomes. How can such a pathway be elucidated? Here we concentrate on the formation of connexon structures, the hexameric 'hemi-channels' spanning single membranes. To determine connexon structures the following questions have to be answered: protein sequence and molecular weight of subunits, orientation of subunits in membranes, molecular weight and subunit composition of oligomers, and pathway and kinetics of trafficking to the cell surface. The first step of connexon assembly in endoplasmatic reticulum membranes
(ER membranes) can be studied in an in vitro protein translation system.
These are cell free cytoplasmic extracts of red blood cells enriched
with membrane fractions (microsomes) of ER from dog pancreas. The latter
is specially rich in ER membranes because the main purpose of the pancreatic
cells is the production and secretion of hormones like insulin and proteases
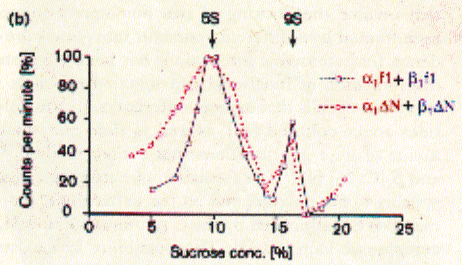
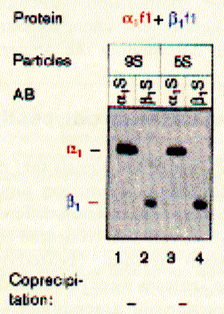
like pepsin. Using sucrose gradient centrifugation, protein particles can be separated according to their size in sucrose solution (see Zonal Sedimentation, chapter 5.2.2 vanHolde). The molecular weight of particles in these centrifugation experiments can be determined by determining the sedimentation coefficient S. Large coefficient indicate large complexes, low S values indicated small complexes. Since density is a measure of mass/volume, a tightly packed protein complex shows a higher density then a single monomer. This is shown in the gradient elution profile above. Radiactive labelled connexin a1 or b1 (and deletion mutants thereof DN) complexes are extracted from their cellular environemnent. Since alpha 1 (43kD) and beta 1 (31kD) connexins have a different molecular weight, they can be sepatated by SDS gel electrophoresis. The western blot analysis (using anti bodies specific against either connexin type) shows that both fraction from the centrifugation contain a mixture of the two connexin types. The electrophoresis analysis, however, does not tell us if they are found as complex or independent units. Sedimentation is analogous to electrophoresis where molecules are moved
within solution at constant speed, pulled by a gravitational force and
opposed by a frictional force of equal but opposite sign. When the gravitational
force is generated in a centriguge, there are three forces acting on
proteins in solution: - a centrifugal force Fc = w2rm
Fc + Fb + Fd = 0 For the mass of solution displaced m, we can substitute the product
of particle mass m, its partial specific volume n(-),
and solution density r. The sedimentation
coefficient S is defined by the velocity of the particel divided by
the centrifugal field strength (equation 1). A small volume of a protein
solution put on top of a sucrose gradient in a centrifuge tube will
behave as a moving boundary along the centrifuge tube axis, with the
boundary broadening over time because of diffusion of the proteins within
solution. Sedimentation coefficients are larger for large particles (see 9S and 5S particle for connexin hexamer and monomer, repsectively). Because the sedimentation behavior is not only a function of size, but also shape, the relation ship between sedimentation coefficient and molecular weight is not staightforward. The Svedberg equation (equation 4) shows how the molecular weight M of a protein can be determined by measuring both S and D of the protein in solution. Idealy, if a protein is globular and not hydrated, a plot of M versus S20,w yields a linear line. In fact, plotting S values of proteins this way is a fast way of determining if they are globular or not. In a sucrose density gradient, the velocity of the moving particle
will continually decrease when moving into an increased sucrose concentration.
This, in fact, allows the separation of macromolecules and complex particles
like membrane of different origin with different lipid to weight ratio
not according to the size, but their apparent density. If the sucrose
gradient at its highest density is higher than the density of any particles
in solution, the velocity will be so slow that the centrifugation separates
particles of different density at equilibrium conditions. Proteomics The dramatic advancement of molecular biology and the high thoughput
sequencing of complete genomes of more than 20 microorganisms and two
eukaryotic organisms (yest and C.elegans), 2D gel electrophoresis has
experienced a dramatic resurgence. Mass spectrometry For whole cell analysis of 2D gels, determining the sequence of protein fragments extracted from individual spots by mass spectrometry is the method of choice. Mass spectrometry determines the charge/mass ratio which is higly significant for molecular structures. The smaller the molecule, the easier the interpretation of mass spectra. This is the main reason why for proteomics research, proteins extracted from 2D gels are fragmented into small peptides of 5-15 amino acids and the sequence including that of modified residues, is quickly determined by correlation algorithms that match charge/mass ratio with potential amino acid combination expected for the observed values. Phosphorylation adds two negative charges and an average of 79.9799 g/mol to the molecular weight of a peptide/protein. Partial sequences can then be pieced together and the longer (partial or complete) protein sequence used to search for homologuous proteins (or genes) using programs such as BLAST at GenBank of the National Center for Biotechnology Information, NCBI. Current techniques of mass spectrometry (van Holde, chapter 5.4) used
for the elucidation of peptide sequences include MALDI (matrix assisted
laser desorption ionization) and ESI (electrospray ionization). In both
cases, peptides are ionized, accelerated in vacuum and the time needed
for them to travel over a specific distance correlated with their charge/mass
ratio.
The accuracy of mass spectrometry is better than for any other technique
(deviation from theoretical molecular weight less than 0.1%). Even though
the mass of large proteins can be assessed accurately, fragmentation
of proteins into small peptides is necessary when using the technique
as a micro sequencing device as mentioned earlier. The possible amino
acid combinations, let alone their sequence, can only be determined
for short sequences by matching the mass/charge ratio with possible
amino acid combinations. The PeptIdent server at Expasy contains
a table
with experimentally determined mass values for all possible amino acids
and common chemical modifications. It is used to identify proteins with
peptide mass fingerprinting data, pI and Mw. Experimentally measured,
user specified peptide masses are compared with the theoretical peptides
calculated for all proteins in SWISS-PROT, making extensive use of database
annotations. 5. Absorption spectroscopy; circular dichroism (CD) Absorption Sepctroscopy: Circular dichroism to study secondary structures of proteins Amino acids are optically active molecules and in a polypeptide are often found as part of regular secondary structures. The frequent occurance of alpha helices and beta sheets in proteins has thus been exploited by measuring the presence of such regularly arranged units from circular dichroism spectra of protein solutions. Although CD measurements are not useful to obtain high resolution structures, but merely secondary structure content of a protein, this information is useful to study the status of protein folds. It can generally be assumed that the absence of any alpha helical or beta strand components indicate the unfolded state of a protein. Dichroism occurs when light absorption differs for different direction of polarized light. Light can be polarized either in a linear way, where the plane of the electric vector is fixed while its amplitude oscillates, or in a circular way, where the plane of polarization of the electric vector is modulated while the amplitude remains constant. The electric vector of circularly polarized light describes a helix which may be right-handed or left-handed. Linearly polarized light will be absorbed maximally when parallel to the transition dipole of the sample molecule, while its amplitude is not affected when the plane orients perpendicular to the transition dipole. Transition refers to the transition between energy levels in a molecular structure of electrons in inner and outer shell, or their vibrational and rotational transitions. A transition has a measurable characteristic of direction, such as the different directions assigned to molecular orbitals, in which we can find electrons. This directionality associated with energy transitions can be exploited spectroscopically for structural information. This direction is called the transition dipole, not to be confused with molecular dipoles or induced dipoles for molecular interactions (see chapter 8.5 and 9.1.5 for details). The energy of a wave particel (photon) of different wave length interacts with different levels of interaction; as a consequence, microwave radiation is used to measure rotational transitions, infrared wave length yield information on vibrational transitions in molecules, while visible light und UV photon energies interact with electrons in the outer shell. This is the range of circular dichroism spectra used to determine secondary structure content of macromolecules. X-ray radiation is absorbed by electronic transitions of the inner atomic shell. A molecule can be excited from a lower to a hihger energy state by the absorption of electromagnetic radiation. Microwaves thus increase the temperature of matter by increasing the rotational energy of molecules. Liquids are specially sensitive to energy absorption in the microwave range because the degree of freedom of individual solvent molecules. The increased rotational energy of water molecules, for example, contributes to their kinetic energy and thus increases the temperature of a cup of coffee. In proteins the aromatic bond structures are of importance in spectroscopy. For protein structure studies we are primarily concerned with backbone conformations. The peptide bond amide group is the dominant chromophore of the polypeptide backbone and has a weak absorption maxima at 220nm and a stronger absorption maxima at 195nm. Circular dichroism makes use of the fact that right and left handed polarized light are absorbed slighly differently in asymmetric molecules. Even though individual amide groups in protein backbones have a symmetric transition dipole, their mutual interaction in highly oriented secondary structures induces asymmetries which translated into circular dichroism spectra (difference of absorption of left and right handed polarized light not zero). Protein secondary structure can be revealed based on their characteristic electronic circular dichroism behavior between 190 and 220nm (see Fig. 10.15 in van Holde for details). Table Electronic circular dichroism pattern for protein secondary structures
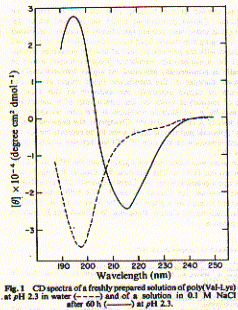
An example of a peptide with a poly-(Valine-Lysine) sequence shows stabilization in beta strand conformation at pH2.3 after being incubated for several hours in 100mM NaCl solution (_____). The initial conformation indicates random coil structure (----). The prominent negative ellipticity at 215nm and a positive band at 195nm is typical for beta sheet formation. Fig. poly (Val-Lys)n electronic CD spectra This figure is taken from a study on peptide with alternating hydrophobic-hydrophilic amino acid residues and their possible significance for prebiotic evolution (A.Brack and L.E. Orgel, 1975, Nature 256,383). A random coil to beta sheet induction could also be achieved by adjusting the pH to 8.8. This transition was reversible and occured within 15 minutes. The peptide precipitated at pH 9.0 indicating the strong propensity of beta strand peptides to form elongated sheet structures. These sheets eventually become insoluble. Such a aggregation mechanism based on beta strand interaction is also suspected to be the major mechanism behind the formation of amyloidogenic plaques such as found in Alzheimer's disease. Similarly, sedondary structure content of larger proteins can easily be determined using CD. This is a particularly useful technique to study folding behavior of proteins such as bacterial porins. These outer membrane proteins are composed of three identical subunit, each of which forms a 16 or 18 stranded anti-parallel beta barrel. CD spectra of porin preparations show typical minima at 215nm. Fig. CD spectra of phosphate selective porin of E.coli
(PhoE; PDB accession number 1PHO)
and PDB structure of related OmpF at 2.4 A resolution (PDB accession
number 2OMF)
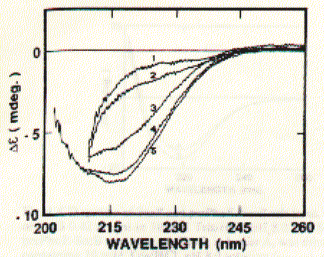
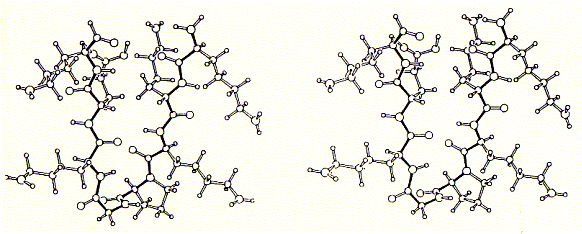
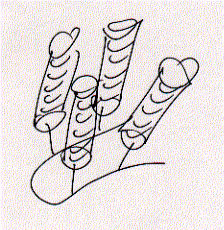
As shown in the figure for phosphoporin of E.coli (PhoE), CD measurements can be used for protein denaturation and refolding studies. Because porins are almost entirly made of beta sheet with less than 3% alpha helical content, their electronic CD spectra are easy to interprete. The figure shows a increase in negative ellipticity at 215nm with decreasing urea concentration. Note that at 8M urea, porin proteins are completely denatured. A synthetic peptide channel approach makes use of alpha helical bundles which are tethered by a short turn forming peptide. Fig. Stereo view of a 4 alpha helical bundel peptide (design
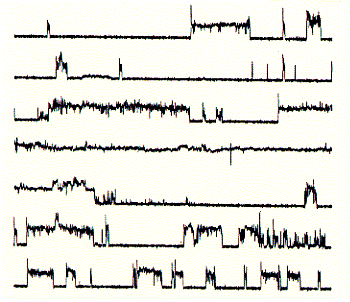
by M.Mutter) The final structure is represented as a cartoon. The horseshooe like
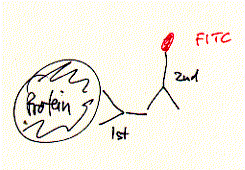
nona-peptide contains four lysine residues. Initial experiments of synthetic peptides which were not covalently liked clearly showed ion channel activity in planar membranes, although the current amplitutes proved to be heterogenous reflecting the intrinsic variality of such a self-assembly channel system. In the covalently linked 4-helical bundle arrangement, a single unitary conductance could be ovbserved representing the ion channel formed by the four alpha helical transmembrane peptides. Comparing channel forming properties from peptides with many different sequences, the amphipathic characteristic of the alpha helix proved to be important, because entirely hydrophobic peptides did not induce any channel like activity, also the membrane stability was compromized indicating peptide membrane interaction. Naturally occuring peptides with many positively charged amino acid residues (magaining peptides from frog skin) are strong antibiotic agents and show membrane disruptive activity rather than controlled stable ion channel activity. Fig. Frog skin antibiotic magaining 2 shows typical Having established an experimental ion channel assay systems allows the measurement of local anaesthetic activity. 4-helical bundle peptides with the sequence from the delta subunit of the muscular nicotinc acetylcholine receptor protein (M2d) can be blocked by the lidocaine derivative QX-222 in the same way as the native receptor channel. 6. Emission spectroscopy; fluorescence, diffusion and distribution GFP - green fluorescence protein Fluorescence labelled antibodies (FITC
- Fluorescein Isothiocyanate - label on secondary antibody, binds to
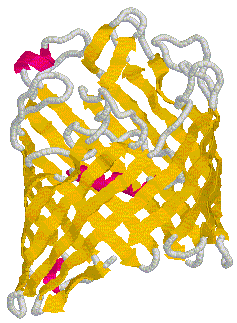
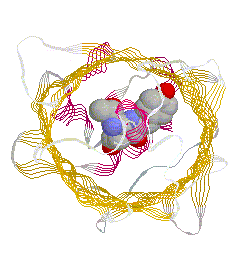
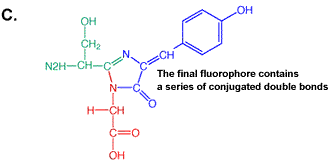
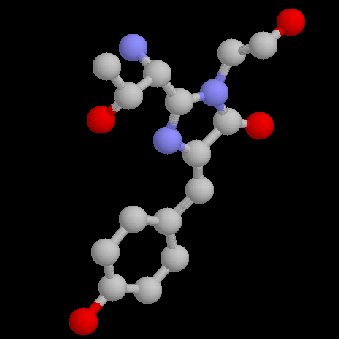
primary antibody which specifically recognized a target protein) have A 236 amino acid, beta barrel forming protein from the jelly fish Aequorea Victoria has revolutionized 'live' imaging of cellular processes. Green fluorescence protein -- GFP -- exhibits fluorescence in the green visible range after being excited by blue light (fluorescence emission wavelength are longer that excitation wavelength). The three consecutive amino acid residues -Ser65-Tyr66-Gly67- inside the beta barrel structure autocatalytically combine to form a chromophore. Fig. GFP protein: side and top view of barrel with central chromophore
at 2.13 Å (1EMB) Fig.Composition of GFP chromophore and rasmol version (from PDB accession 1EMB)
GFP is a 236 amino acid protein with the following sequence: >1emb_ mol:protein-het length:236 Green Fluorescent Protein
(Gfp) The X in the sequence above indicates the chromophore position. Fluorescence, general Fluorescence occurs when excited electrons in a singlet state (electron
spins in molecule are EX (to 2nd or higher singlet) excitation - excitation state lifetime
- emisson (from 1st singlet) EM While absorption is very fast and takes about 10-15 seconds, internal conversion to the first singlet level is about 1,000 fold slower. The actual lifetime of the electron in the first excited singlet, before fluorescence emission occurs, is fairly long and lasts about 10-8 seconds. Fluorescence is a spontaneous process and is favoured in conjugated double bond systems of the pp* transition, because the probability of a spontaneous emission is related to the integrated intensity of an absorption band; the stronger the intensity of absorption of the first singlet, the higher the probability for fluorescence (the energy of a transition is determined by the spacing between the energy levels; however, not all photons of the correct energy quanta will be absorbed, the fraction of photons absorbed is the intensity of absorption; see vanHolde chapters 8.4 and 11 for details). This is found in sidechains of tryptophane and tyrosine amino acids and is more pronounced in the autocatalytically derived chromophore sidechain of GFP. The entire wavelength of visible light results in a fluorescence spectra,
either absorption or emission spectra. Excitation of a fluorophore
at three different wavelengths (EX 1, EX 2, EX3) does not change the
emission profile but does produce variations in fluorescence emission
intensity (EM 1, EM 2, EM 3) that correspond to the amplitude of the
excitation spectrum. Quenching Since loss of energy of an excited electron can occur as radiation
(fluorescence) and non-radiation process (internal conversion, heat),
the quantum yield of a substance is the ratio of the total number of
quanta emitted to the total number of quanta absorbed. The quantum yield
can be related to the loss of fluorescence over time, or fluorescence
decay, which follows a first-order process, like radioactivity.
In other words, the fluorescence events of each molecule is independent
of its neighbors. The number of molecules N(t) that still can emitt
fluorescence after time t is given as: N(t) = N(0)
e-kt By definition, the lifetime t of an ensemble of N fluorescent molecules is 1/k. The lifetime can be measured because the intensity is proportional to the number N of molecules in the excited state at time t. The lifetime is simply the time it takes for the maximum intensity to decrease by the factor 1/e. The quantum yield differes with the molecular environment because this
determines the potential non-radiation loss of energy. The quantum yield
of a fluorophore often increases when the molecule is taken up from
solution and binds to DNA or proteins. Thus, measuring quantum yields
is indicative of binding. It is not necessary to know the exact quantum
yield in order to use fluorophores for biochemical studies. The intensity
of the fluorescence spectra changes with changing environment (solvent,
ionic strength, lipid environment, pH etc.) and is referred to as quenching
if the intensity decreases. The loss of intensity will depend on fluorophore
concentration, shape of fluorescence spectrum and the wavelength chosen
for observation (intensities vary differently at different wavelength).
This is of course reminiscent of electronic CD spectroscopy. Energy transfer A special form of quenching is the energy transfer between fluorescent molecules, where a donor activates electrons in an acceptor molecule. This interaction is a function of the distance between donor and acceptor and can be exploited to study conformational flexibility in macormolecues. The distance dependency follows an r-6 function and thus is a short range effect. Requirements for energy transfer are transition dipole interaction between the two fluorophores and an overlap of the fluorescence spectrum of the donor with the absorption spectrum of the acceptor. Photobleaching Under high-intensity illumination conditions, the irreversible destruction or photobleaching of the excited fluorophore becomes the factor limiting
|

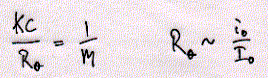

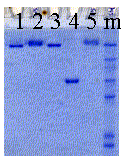 For
most proteins, subunit interaction is not strong enough to withstand
the denaturing effect of SDS. Bacterial porins, however, show an unusual
resistance toward SDS denaturation, including a strong association with
the bacterial cell wall glycolipid lipopolysaccharide (LPS). The small
gel shown here shows porin trimers in association with different amounts
of the glycolipid LPS. Control lane 1 shows LPS-free porin trimers,
while the sample in lanes 2 and 5 contains porin trimers associated
with native LPS from E.coli outer membranes. Lane 3 shows a porin trimer
with a synthetic, short chain LPS exhibiting a low affinity. This lipid
A type of lipopolysaccharide dissociates from porin in SDS solution.
Lane 4 shows a heat denaturated monomer. The banding pattern in the
right lane (m) contains the molecular marker proteins as described above.
The lower mobility of some porin trimers (banding pattern with higher
molecular weight) is due to the binding of different amount of lipopolysaccharide
units. The ratio of glycolipid to trimer is not well defined and varies
from sample to sample. Heat denaturation, however, removes any residual
glycolipids from porin monomers. Monomers never show a banding pattern
as do the native trimers. Below is a schematic representation of the
cell wall composition of E.coli showing major inner and outer membrane
components.
For
most proteins, subunit interaction is not strong enough to withstand
the denaturing effect of SDS. Bacterial porins, however, show an unusual
resistance toward SDS denaturation, including a strong association with
the bacterial cell wall glycolipid lipopolysaccharide (LPS). The small
gel shown here shows porin trimers in association with different amounts
of the glycolipid LPS. Control lane 1 shows LPS-free porin trimers,
while the sample in lanes 2 and 5 contains porin trimers associated
with native LPS from E.coli outer membranes. Lane 3 shows a porin trimer
with a synthetic, short chain LPS exhibiting a low affinity. This lipid
A type of lipopolysaccharide dissociates from porin in SDS solution.
Lane 4 shows a heat denaturated monomer. The banding pattern in the
right lane (m) contains the molecular marker proteins as described above.
The lower mobility of some porin trimers (banding pattern with higher
molecular weight) is due to the binding of different amount of lipopolysaccharide
units. The ratio of glycolipid to trimer is not well defined and varies
from sample to sample. Heat denaturation, however, removes any residual
glycolipids from porin monomers. Monomers never show a banding pattern
as do the native trimers. Below is a schematic representation of the
cell wall composition of E.coli showing major inner and outer membrane
components. 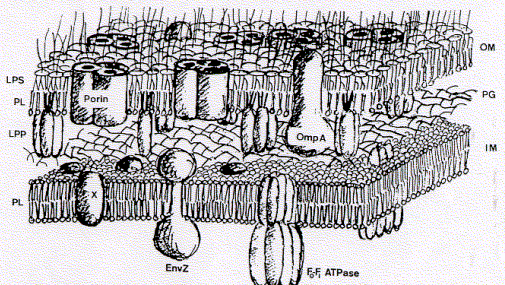
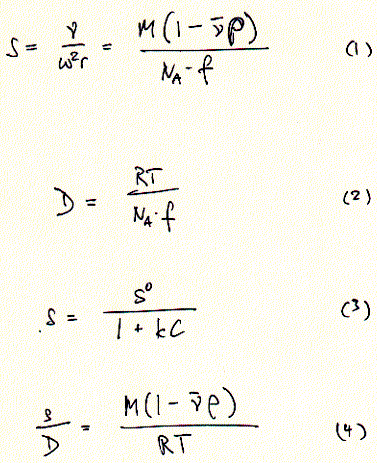 The
definition of the diffusion coefficient is given in equation 2 indicating
that diffusion is proportional to the kinetic energy of the particle
and inversely proportional to the frictional coefficient. Zonal sucrose
gradient centrifugation minimizes the inpact of diffusion making the
moving boundary more stable. If the protein solution contains a mixture
of protein complexes of different size, multiple moving boundaries will
form after several hours of running the centrifuge. In addition, the
boundary zone for large particle will be sharper because diffusion is
much smaller due to the larger frictional forces of the large particles
as compared to smaller ones. Although the sedimentation coefficinet
is a characteristic (intrinsic) property of each protein, nucleic acid,
polysaccharide etc., it depends on the temperature and ionic strength
of the solution. Experimental measurements of sedimentation coefficient
S therefore are related to standard conditions, for which a characteristic
S20,w has been determined (tabulated
in many references). Finally, S will also depend on the particel concentration
because particles may interact with each other resulting in an apparent
increased frictional coefficient. When f0 is the frictional
coefficient at concentration zero (ideal solution, no particle interaction),
the effective sedimentation coefficient S is determined as indicated
by equation 3 (with S0 the sedimentation coefficient for
an ideal, diluted solution at C = 0).
The
definition of the diffusion coefficient is given in equation 2 indicating
that diffusion is proportional to the kinetic energy of the particle
and inversely proportional to the frictional coefficient. Zonal sucrose
gradient centrifugation minimizes the inpact of diffusion making the
moving boundary more stable. If the protein solution contains a mixture
of protein complexes of different size, multiple moving boundaries will
form after several hours of running the centrifuge. In addition, the
boundary zone for large particle will be sharper because diffusion is
much smaller due to the larger frictional forces of the large particles
as compared to smaller ones. Although the sedimentation coefficinet
is a characteristic (intrinsic) property of each protein, nucleic acid,
polysaccharide etc., it depends on the temperature and ionic strength
of the solution. Experimental measurements of sedimentation coefficient
S therefore are related to standard conditions, for which a characteristic
S20,w has been determined (tabulated
in many references). Finally, S will also depend on the particel concentration
because particles may interact with each other resulting in an apparent
increased frictional coefficient. When f0 is the frictional
coefficient at concentration zero (ideal solution, no particle interaction),
the effective sedimentation coefficient S is determined as indicated
by equation 3 (with S0 the sedimentation coefficient for
an ideal, diluted solution at C = 0).  The
new science of proteomics addresses the protein expression pattern of
cells to understand the response of cells or organisms for certain signals
-- hormonal, neurobiological, immunological, or developmental. Although
mRNA levels are normally used to assess the presence of proteins by
studying the gene expression levels (Northern blot analysis), many times
the concentration of mRNA poorly correlates with actual levels of proteins
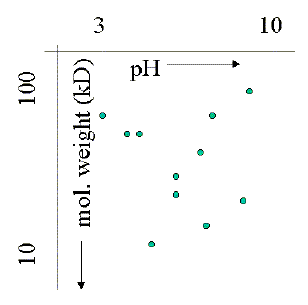
in a cell. The image on the left is taken from a 2D gel of a plant cell
extract from Arabidopsis thaliana. As the sample gel shows, and this
is only 10% of the entire pH and molecular weight range on the
The
new science of proteomics addresses the protein expression pattern of
cells to understand the response of cells or organisms for certain signals
-- hormonal, neurobiological, immunological, or developmental. Although
mRNA levels are normally used to assess the presence of proteins by
studying the gene expression levels (Northern blot analysis), many times
the concentration of mRNA poorly correlates with actual levels of proteins
in a cell. The image on the left is taken from a 2D gel of a plant cell
extract from Arabidopsis thaliana. As the sample gel shows, and this
is only 10% of the entire pH and molecular weight range on the 
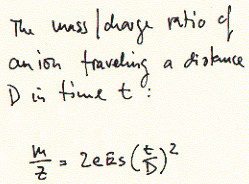
 A single
alpha helix is synthesized to the side chain NH2 group forming four
parallel arranged peptides with equal sequence. The amino acid sequences
can be choosen at will and are representing the channel ligning transmembrane
sequences of the nicotinic acetylcholine receptor (M2) or Ca selective,
voltage gated ion channel subunit alpha.
A single
alpha helix is synthesized to the side chain NH2 group forming four
parallel arranged peptides with equal sequence. The amino acid sequences
can be choosen at will and are representing the channel ligning transmembrane
sequences of the nicotinic acetylcholine receptor (M2) or Ca selective,
voltage gated ion channel subunit alpha.
 been
used for a long time to show the cellular localization of proteins.
Also the addition of smaller fluorescence labels has been employed that
reversibly bind to macromolecules or can be chemically cross linked
to demonstrate the surface exposure of certain amino acid residues.
Recently, autofluorescent proteins have been used through recombinant
DNA technology to avoid the often chemically harsh treatment of cells
neede to bring fluorescence labels into subcellular compartments. Here,
protein chimeras are made where a protein 'gains' a domain which carries
a label which can easily observed by light microscopy. One very successfull
label protein is the green fluorescent protein, or GFP.
been
used for a long time to show the cellular localization of proteins.
Also the addition of smaller fluorescence labels has been employed that
reversibly bind to macromolecules or can be chemically cross linked
to demonstrate the surface exposure of certain amino acid residues.
Recently, autofluorescent proteins have been used through recombinant
DNA technology to avoid the often chemically harsh treatment of cells
neede to bring fluorescence labels into subcellular compartments. Here,
protein chimeras are made where a protein 'gains' a domain which carries
a label which can easily observed by light microscopy. One very successfull
label protein is the green fluorescent protein, or GFP. 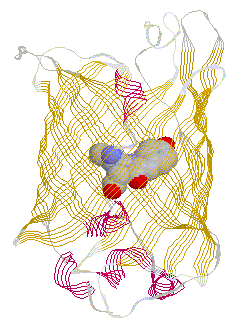
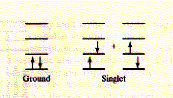 paired;
see orientation of arrows in picture), after absorption of light quanta,
fall back into their ground state and emitt themselves light in the
visible range. The wavelength of the emitted light is usually longer
than that of the activating light because of internal loss of energy
before fluorescence occurs. This phenomenon is called the Stoke's shift
and refers to the loss of energy during the excited state of the electron
(no radiation) where internal conversion brings the excited electron
to the lowest (first) level singlet.
paired;
see orientation of arrows in picture), after absorption of light quanta,
fall back into their ground state and emitt themselves light in the
visible range. The wavelength of the emitted light is usually longer
than that of the activating light because of internal loss of energy
before fluorescence occurs. This phenomenon is called the Stoke's shift
and refers to the loss of energy during the excited state of the electron
(no radiation) where internal conversion brings the excited electron
to the lowest (first) level singlet.  fluorescence
detectability. Recent investigations have provided a detailed description
of the multiple photochemical reaction pathways responsible for photobleaching
of fluorescein (excitation/emission maxima ~494/520 nm;
fluorescence
detectability. Recent investigations have provided a detailed description
of the multiple photochemical reaction pathways responsible for photobleaching
of fluorescein (excitation/emission maxima ~494/520 nm; {kind=link}